

The Tree of Life: Tangled Roots and Sexy Shoots
Tracing the genetic pathway from the Last Universal Common Ancestor to Homo sapiens
Chris King Dec 2009 - Jul 2024 Genotype: 1.5.41
PDF printable version (With hi-res images. Print in Chrome to PDF)
For significant updates, follow @dhushara on Twitter
Abstract: This article is a fully referenced research review to overview progress in unraveling the details of the evolutionary Tree of Life, from life's first occurrence in the RNA-era, to humanity's emergence and diversification, through migration and intermarriage.The Tree of Life, in biological terms, has come to be identified with the evolutionary tree of biological diversity. It is this tree which represents the climax fruitfulness of the biosphere and the genetic foundation of our existence, embracing not just higher Eucaryotes, plants, animals and fungi, but Protista, Eubacteria and Archaea, the realm, including the extreme heat and salt-loving organisms, which appears to lie almost at the root of life itself. The notion of a tree of evolution veertically down te generations has become complicated by evidence for promiscuous horizontal gene transfer and for genetic symbiosis at the root of the eucaryote tree. This review will cover all these aspects, from the first life on Earth to Homo sapiens.
Prequel: Biocosmology a definitive overview of the research to elucidate the origin of life on Earth and to establish life as an interactive manifestation of cosmic symmetry-breaking.
Contents
Fig 0: Comprehensive Evolutionary Tree of Life (King) Click on images to enlarge.
See also:
Introduction: The Comprehensive Tree
Linked in figure 1 is a high-resolution image of the evolutionary tree of life, from viruses through bacteria and archaea to protista, plants, animals and fungi, with a selection of representative species illustrated. I have updated and amended this several times as new research has clarified specific parts of the trunk and branches. The evolutionary tree of life is our immortal progenitor, not just of ourselves, but of all the species with which we co-depend, so we need to both understand it and protect it for the future generations. This initial tree forms a good representation of the evolution of higher plants and fungi, so the remainder of the article will examine the tortuous route from the last common ancestor, through the eucaryotes to metazoa, and ultimately to humanity, language and culture.
This article seeks to be a real time account of the discovery processes showing us in ever-incteasing detail, the nature of the tree and its many tangled interactions, both at the genetic and organismic level. It also strives to be a fully up-to-date scientfic account of the discovery process for which we all owe a vote of thanks to the many researchers whose work is illustrated and cited in this extensive review article.
Where the trees are complicated and detailed, high-resolution versions can be viewed by clicking each of the images. A high-resolution PDF version is also provided.
LUCA: The Last Universal Common Ancestor

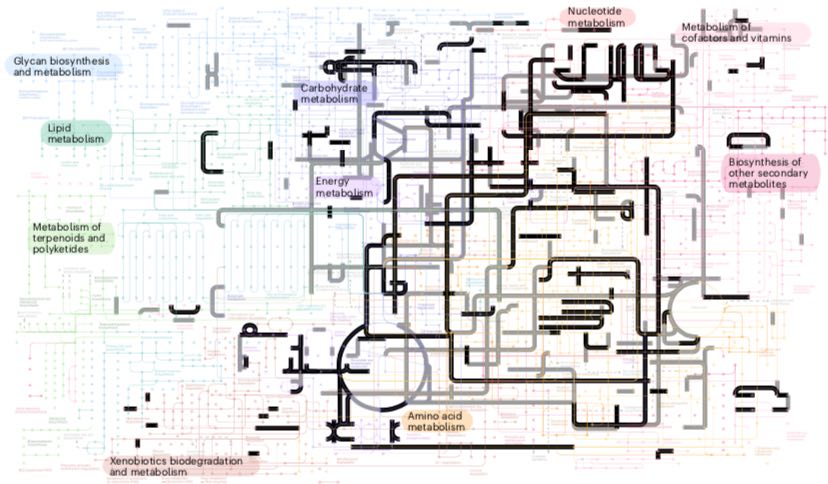
Fig 1: Early origin of LUCA around 4.3 bya, and ensuing last common ancestors of archaea, bacteria and eucaryotes. Right LUCA metabolic pathways (Moody et al. 2024).
Following a phase of biogenesis possibly emerging directly from cosmic symmetry-breaking (King 1978, 2004), based on spontaneous prebiotic RNA synthesis (Powner et. al. 2009, 2010) recent research suggests that the last universal common ancestor (LUCA) of all life on the planet may have arisen before the first cells, from a phase interface between alkaline hydrogen-emitting undersea vents and the archaic acidified iron-rich ocean (Martin and Russel 2003) in which differential dynamics in membranous micropores in the vents managed to concentrate polypeptides and polynucleotides to biologically sustainable levels (Baaske et. al. 2007, Budin et. al. 2009), giving rise to the RNA era, while at the same time providing a free energy source based on proton transport across membranous microcellular interfaces resulting from fatty acids also being concentrated above their critical aggregate concentration. 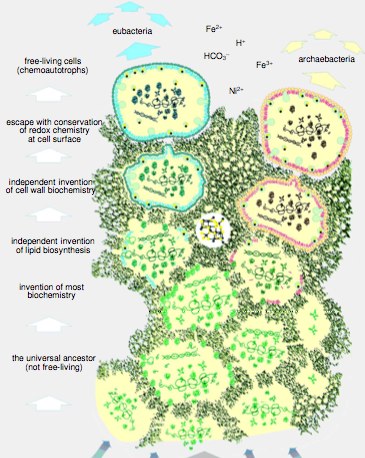 The transition to enclosed cells is likely to have been in an active iron-sulphur reaction phase still present in living cells and associated with sodium-proton anti-porters activating ATP (Lane and Martin 2012, Lane 2009b), leading in turn to electron transport and some of the most ancient proteins, such as ferredoxin,
The transition to enclosed cells is likely to have been in an active iron-sulphur reaction phase still present in living cells and associated with sodium-proton anti-porters activating ATP (Lane and Martin 2012, Lane 2009b), leading in turn to electron transport and some of the most ancient proteins, such as ferredoxin,
Fig 1a: Proposed scheme for the universal common ancestor (Martin and Russel 2003)
The universal common ancestor of the three domains of life may have thus been a proton-pumping membranous interface from which archaea and bacteria emerged as free-living adaptions. This is suggested by fundamental differences in their cell walls and other details of evolutionary relationships among some of the oldest genes.
Although it has been suggested that glycolysis evolved before ion pumping and electron transport (Alberts et al. 2002, Koonin 2003), respiratory electron transport is universal to the three domains of life including eucaryote mitochondria and chloroplasts and both bacteria and archaea (Schafer et al. 1996, 1999, see fig 1d). Among the archaea, halobacteria still use a form of photosynthesis generating ATP from H+ gradients generated by a rhodopsin protein and those in hydrothermal vents rely on Na+-H+ antiporters to generate ion gradients, and their membrane proteins, such as the ATP synthase, are compatible with gradients of sodium ions or protons (Lane and Martin 2012, Yong 2012). The archaea also use a unique form of electron transport in methanogenesis (Schafer 2004).
 Fig 1b1: (Above) founding metabolism based on Na+-H+ anti-transported, ATP synthetase and FeSNiS containing vents (Lane and Martin 2012). The extremely ancient origin of the rhodopsin family of heptahelical receptors can be seen from the ultra-primitive archael photosynthesis in Halobacteria, which relies on direct coupling between photo-stimulated chemiosmotic H+ pumping and H+ generated ATP formation, based on bacteriorhodopsin, which is heptahelical, uses a form of retinal and whose helices may share a distant sequence homology with vertebrate rhodopsin (Pardo et al, Taylor & Agarwal, Soppa, Ihara et al, Shen et al.) (click to enlarge).
Fig 1b1: (Above) founding metabolism based on Na+-H+ anti-transported, ATP synthetase and FeSNiS containing vents (Lane and Martin 2012). The extremely ancient origin of the rhodopsin family of heptahelical receptors can be seen from the ultra-primitive archael photosynthesis in Halobacteria, which relies on direct coupling between photo-stimulated chemiosmotic H+ pumping and H+ generated ATP formation, based on bacteriorhodopsin, which is heptahelical, uses a form of retinal and whose helices may share a distant sequence homology with vertebrate rhodopsin (Pardo et al, Taylor & Agarwal, Soppa, Ihara et al, Shen et al.) (click to enlarge).
The H+-dependent ATPsynthase universal to the chemiosmotic coupling of electronand ion transport to ATP production is a rotary motor which appears to have evolved from two separate subunits, one of which has been proposed to be a helicase (Doering et al. 1995, Crofts 1996). Hexameric helicases are found both in the SF3 superfamily in viruses (Hickman & Dyda 2005) and the MCM helicases are critical to replication forks in diverse organisms from humans to archaea (Fletcher et al. 2003), Onesti & MacNeill 2013, Sharma et al. 2006). The viral SF3 superfamily (Leitão 2015) helicase tree shows variants active on both RNA and SNA substrates, consistent with an origin in the RNA era (Caprari et al. 2015). Supporting the notion of subunits, a beta-chain of ATP synthase is homologous to a hepatic lipoprotein receptor (Martinez et al. 2003).
 Fig 1b2: Left: Rotary action of ATPsynthase, shown centre. See video.
Fig 1b2: Left: Rotary action of ATPsynthase, shown centre. See video.
Respiratory electron transport occurs in both aerobic and anaerobic organisms and the terminal oxidases, iron-sulphur proteins and flavin-binding polypeptides all show evolutionary trees reaching back to the common ancestor of the three domains, implying terminal oxidases predate oxygenic photosynthesis. The fact that many components of archaeal electron transport are significantly different in structure from those of bacteria implies these evolved separately and that archaeal electron transport is not simply a more recent result of horizontal transfer (Schafer 2004). Terminal oxidases belonging to oxygen, nitrate, sulfate, and sulfur respiratory pathways have been sequenced in members of both bacteria and archaea including cytochrome oxidase, nitrate reductase, adenylylsulfate reductase, sulfite reductase, and polysulfide reductase which can likewise be assigned to LUCA (Castresana & Moreira 1999). Similar considerations apply to ferredoxins, one of the most ancient coded proteins (Fitch & Bruschi 1987, Hall, Cammack & Rao 1974).

Fig 1b2a: (1) The H+-dependent ATPsynthase universal to the chemiosmotic coupling of electron and H+ ion transport to ATP production is a rotary motor which appears to have evolved from two separate subunits F0 and F1 (Mahendrarajah et al. 2023). The F1 subunit is purported to have descended from an ATP-dependent helicase, and the F0 from a passive ion channel
(Nirody, et al. 2020). F1 unit has independently been proposed to share ancient structure with a polynucleotide helicase (Doering et al. 1995, Gomis-Rüth et al. 2001). Hexameric helicases are found both in the SF3 superfamily in viruses and the MCM helicases are critical to replication forks in diverse organisms from humans to archaea. The viral SF3 superfamily helicase tree shows variants active on both RNA and DNA substrates, consistent with an origin in the RNA era. (Caprari et al. 2015).
 Fig 1b2b: Cloverleaf secondary structure representation of tRNA showing post-transcriptional nucleoside modifications that are conserved among bacteria and archaea in both identity and position. The structures of respective conserved modified nucleosides are highlighted in grey. Methyl and acetyl groups are shown in red and dark red, respectively; sulfur in yellow; and the threonylcarbamoyl group in blue (Weiss et al. 2018).
Fig 1b2b: Cloverleaf secondary structure representation of tRNA showing post-transcriptional nucleoside modifications that are conserved among bacteria and archaea in both identity and position. The structures of respective conserved modified nucleosides are highlighted in grey. Methyl and acetyl groups are shown in red and dark red, respectively; sulfur in yellow; and the threonylcarbamoyl group in blue (Weiss et al. 2018).
Transfer RNA requires modified bases for proper interaction with mRNA (codon–anti-codon wobble base pairing) and with rRNA in the ribosome during translation. That is, modi- fied bases are part of the universal genetic code (Fig 4), which was present in LUCA. Many RNA-modifying enzymes trace to LUCA, particularly the enzymes that modify tRNA. Several of those enzymes are methyltransferases (many SAM dependent), and they remind us that, before the genetic code arose, the four main RNA bases could hardly have been in great supply in pure form because there were no genes or enzymes, only chemical reactions..
 Fig 1b3: Evolutionary trees for two components of the electron transport chain, Fe-S proteins (left) and flavin-binding polypeptides (right archaea lower right Homo sapiens upper left), span the three domains of life (Schafer et al. 1996, Schafer 2004).
Fig 1b3: Evolutionary trees for two components of the electron transport chain, Fe-S proteins (left) and flavin-binding polypeptides (right archaea lower right Homo sapiens upper left), span the three domains of life (Schafer et al. 1996, Schafer 2004).
It has also been proposed, on the basis of the highly-conserved commonality of transcription and translation proteins to all life, but the apparently independent emergence of distinct DNA replication enzymes in archaea/eucaryotes and eubacteria, that the last universal common ancestor had a mixed RNA-DNA metabolism based on reverse transcriptase, pinpointing it to the latter phases of the RNA era (Leipe et. al. 1999).
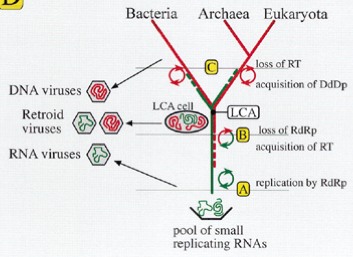 Fig 1b4: Hypothetical branching and evolution of RNA and DNA replication machinery (Leipe et. al. 1999) suggests viruses were pivotal in the transition from RNA to DNA (see below)
Fig 1b4: Hypothetical branching and evolution of RNA and DNA replication machinery (Leipe et. al. 1999) suggests viruses were pivotal in the transition from RNA to DNA (see below)
To get a characterization of LUCA at the point it diversified into the three domains of life Archaea, Eucaryotes and Bacteria, one cannot rely on nucleotide gene sequences because these would have mutated beyond recognition, but amino acid sequences mutate more slowly because neutral mutations leave the amino acid sequence fixed and the tertiary folded structure of a protein is even more strongly conserved.
The validity of the RNA-era concept and the capacity for RNAs to be both replicating informational and active ribo-enzymes is emphasized by the continuing dependence of the ribosome on rRNA rather than the protein components demonstrated by the 3D realizations of the two subunits in fig 1c1, which show that the rRNA molecules are still carrying out the central task of protein assembly with only minor modification due to the 'chaperoning' proteins, despite 3.8 billion years of evolution.


Fig 1c1: Small and large rRNA subunits of the eubacteia Thermus thermophilus and the archaeon Haloarcula marismortui.
RNA orange and yellow, protein blue and active site green. (Wikipedia Ribosome) Click the image to see the RNAs rotating.
Brooks et al. (2002) have found that the amino acids used in sections of genes common to life which are believed to originate with LUCA show amino acid distributions reflecting the relative abundance of such amino acids in primitive synthesis, indicating that the first translational genes used the amino acids which were spontaneously available, consistent with my original hypothesis on origin of the genetic code in Biocosmology. A specfic model of the evolution of the ribosome envisages that the smaller subunit which binds to and moves along the mRNA began first as an RNA-based RNA helicase which was essential to avoid the RNA era ending in non-replicating double stranded hairpins (Zenkin 2012). This would have then coupled to the larger subunit which could have assembled transfer RNAs coupled to amino acids via ribozymes, resulting in a simple genetic code, for example based on polar and non-polar amino acids.
It is also possible to investigate aspects of the genetic landscape before LUCA. In particular, in the evolution of the aminoacyl-tRNA synthetases (aaRS), coupling an amino acid to its respective t-RNA, analysis of genetic trees shows that there have been multiple horizontal transfer of such genes, including some from putative sister species of LUCA, in a similar manner to the introgression of Neanderthal DNA into Homo sapiens, as well as evolutionary diversification of these enzymes from common ancestors with more generic amino acid affinities.
A pivotal idea that is central to the establishment and evolution of the genetic code is the feedback loop through t-RNA synthases that is essential for the ribosome to be able to encode proteins via their t-RNAs. The synthases come in two classes, which have completely different protein structures and also couple onto the t-RNA in different ways, involving both the 2OH vs 3OH attachment sites in the terminal adenosine and the major or minor groove involved in the approach. The Rodin-Ohno (1995) hypothesis that these two classes originated from complementary sense and anti-sense transcripts of the same gene has received experimental support from studies (Martinez-Rodriguez et al. 2015, Carter 2017) showing that when the enzymes are pruned down to their evolutionary core they remain catalytic by several order of magnitude. This provides convincing evidence for a primal origina from a single gene able to catalyse distinct classes of amino acid by distinct processes. These may have both initially been involved together facilitating overall coupling (de Pouplana 2020), before adopting complementary roles as their enficiency increased. Carter & Wills (2018, Wills & Carter 2018) have established routes of viable establishement of the underlying feedback loops enabling the emergence of amino-acid selectivity.

Fig 26b2: (a) Possible evolution of t-RNA synthases from a complementary pair of sense and antisense enzymes (de Pouplana(2020). (b) The Rodin-Ohno (1995) hypothesis of paired sense-antisense transcripts leading to class 1 & 2 synthases. (c, e) The complementary structures of the two classes and their amino acid assignments (Kaiser et al. 2018). (d) Overall structures (Wikipedia) (f) The complementary enzymes retain catalytic activity when reduced to a 49 amino acid evolutionary core based on complementary DNA strands (Martinez-Rodriguez et al. 2015). (g) Evolutionary processes driving formation of the genetic code (Carter 2017). (h) tRNA acceptor-stem bases considered in the analysis of aaRS groove recognition (Carter & Wills 2018).
 Fig 1c1b: Differences between archaeal and bacterial/eucaryote membranes.
Fig 1c1b: Differences between archaeal and bacterial/eucaryote membranes.
One intriguing indication of the state of genetic translation in LUCA is the incorporation of selenocysteine into the genetic code. Selenoenzymes which contain selenocysteine as a genetically translated amino acid are essential to the three domains of life and source back to LUCA, despite the fact that the 21st coded amino acid selenocysteine could not be fitted into the genetic code. An ingenious piece of genetic software engineering evolved in which the amber stop codon UAG is overridden if the m-RNA possesses a motif called SECIS (selenocysteine insertion sequence). Selenocysteine is then inserted instead of termination, and translation continues. 
Fig 1c2: Left: Evolutionary tree of selenophosphate synthetase (Romero et al. 2005) spans the three domains of life. Centre: SECIS hairpins of archaea (A), bacteria (B) and corresponding eukaryote variants (C, D) (Moldave ed 2006). Top right: Tertiary structure of SECIS showing highly conserved regions (hot) (Walczak et al. 1996). Lower right: SECIS acts as an RNA-enzyme to attach the selenocysteine t-RNA to the nascent protein (click to enlarge).
SECIS is an unusual hairpin loop structure which has varying forms in archaea and prokaryotes with both forms appearing in eucaryotes, but they have a common feature of a highly conserved hairpin loop forming an RNA translational catalyst, which literally takes over some of the ribosomal RNA function, binding to the selenocysteine t-RNA and coupling selenocysteine to the nascent protein chain, as shown in fig 1c2. It is clear that this unique piece of genetic software engineering evolved in LUCA because the wobble positions of three other essential amino acid t-RNAs, lysine, glutamine and glutamic acid (those with two wobble positions XAA-XAG, the fourth set being amber and ochre stop codons), all depend on a modified 2-seleno-uridine base to function and this has to be generated from selenophosphate, which in turn is generated by selenophosphate synthetase. As shown above left, this enzyme has an evolutionary tree extending back to LUCA confirming the obvious - that the genetic code cannot exist without the 21st software engineered amino acid selenocysteine!
In a ground-breaking project to identify genes that can illuminate the biology of LUCA, a team associated with Martin, (Weiss et al. 2016) took a phylogenetic approach to decoding the LUCA metabolism. Among proteins encoded in sequenced prokaryotic genomes, they sought those that: (1) are present in at least two higher taxa of bacteria and archaea, and (2) its tree should recover bacterial and archaeal monophyly. Genes meeting both criteria are unlikely to have undergone transdomain lateral gene transfer (LGT), and thus were probably present in LUCA and inherited within domains since then. By focusing on phylogeny rather than universal gene presence, they identified genes involved in LUCA's physiology - the ways that cells access carbon, energy and nutrients from the environment for growth.
 Fig 1c3: Earliest fossil evidence for LUCA. (a) Lost city hydrothermal vents provide biogenic redox reactions from the interaction of cosmically abundant alkaline olivine(c) with acidic ocean water due to dissolved CO2 to form a crustal chemical garden. These contain fizzing pores (b) generating H2 and producing organics, which are capable of concentrating resulting polymers to biological concentrations (Baaske et al. 2007). Schopf (1993) found 3.5 billion-year-old fossils resembling strings of microscopic cells (d) lying near remnants of 3.6 byrs old stromatolites (e), microbial mats of cyanobacteria, as illustrated at Shark Bay Australia (i). Schopf et al. (2017) analysed these rocks in greater chemical detail by secondary ion mass spectroscopy (SIMS) from a thin section of the 3.465 billion year old Apex chert of northwestern Western Australia, and show their δ13C compositions vary systematically taxon to taxon from 31% to 39%. The SIMS data for the two highest δ13C Apex taxa are consistent with those of extant phototrophic bacteria; those for a somewhat lower δ13C taxon, with nonbacterial methane-producing Archaea; and those for the two lowest δ13C taxa, with methane-metabolizing γ-proteobacteria. The diversity of these, consistent with the existent tree of life, imply life must have originated 500 million years earlier or around 4 billion years ago. Nutman et al. (2016) discovered putative stromatolites (f) colonies of photosdating to 3.7 billion years in the Isua formation, Greenland. Wacey et al. (2011) have found clusters of putative sulphur-metabolizing cells (g) in 3.4-billion-year-old rocks of Western Australia. Dodd et al. (2017) found carbon tube structures from fossil remains of ancient hydrothermal vents dated to 3.7-4.2 billion years. The earliest evidence of life comes from disordered graphite inclusions of zircons from Western Australia, with a high 12C content, consistent with a biogenic origin, that formed 4.1 billion tears ago (Bell et al. 2015). This date is highly significant, since the oldest direct evidence for the presence of surface waters are slightly younger ca. ~3.8 billion years old sedimentary rocks called banded iron formation (BIF) that are exposed at Isua in southwest Greenland.
Fig 1c3: Earliest fossil evidence for LUCA. (a) Lost city hydrothermal vents provide biogenic redox reactions from the interaction of cosmically abundant alkaline olivine(c) with acidic ocean water due to dissolved CO2 to form a crustal chemical garden. These contain fizzing pores (b) generating H2 and producing organics, which are capable of concentrating resulting polymers to biological concentrations (Baaske et al. 2007). Schopf (1993) found 3.5 billion-year-old fossils resembling strings of microscopic cells (d) lying near remnants of 3.6 byrs old stromatolites (e), microbial mats of cyanobacteria, as illustrated at Shark Bay Australia (i). Schopf et al. (2017) analysed these rocks in greater chemical detail by secondary ion mass spectroscopy (SIMS) from a thin section of the 3.465 billion year old Apex chert of northwestern Western Australia, and show their δ13C compositions vary systematically taxon to taxon from 31% to 39%. The SIMS data for the two highest δ13C Apex taxa are consistent with those of extant phototrophic bacteria; those for a somewhat lower δ13C taxon, with nonbacterial methane-producing Archaea; and those for the two lowest δ13C taxa, with methane-metabolizing γ-proteobacteria. The diversity of these, consistent with the existent tree of life, imply life must have originated 500 million years earlier or around 4 billion years ago. Nutman et al. (2016) discovered putative stromatolites (f) colonies of photosdating to 3.7 billion years in the Isua formation, Greenland. Wacey et al. (2011) have found clusters of putative sulphur-metabolizing cells (g) in 3.4-billion-year-old rocks of Western Australia. Dodd et al. (2017) found carbon tube structures from fossil remains of ancient hydrothermal vents dated to 3.7-4.2 billion years. The earliest evidence of life comes from disordered graphite inclusions of zircons from Western Australia, with a high 12C content, consistent with a biogenic origin, that formed 4.1 billion tears ago (Bell et al. 2015). This date is highly significant, since the oldest direct evidence for the presence of surface waters are slightly younger ca. ~3.8 billion years old sedimentary rocks called banded iron formation (BIF) that are exposed at Isua in southwest Greenland.
The presence of the thermophile-specific enzyme reverse gyrase implies that LUCA was a thermophile. A rotator-stator ATP synthase subunit suggests LUCA was able to harness ion gradients for energy. LUCA also appears to have had a gene for a 'revolving door' protein that could swap sodium and hydrogen ions across this gradient. Earlier studies by Martin and Nick Lane of University College London suggest that such a protein would have been absolutely crucial for exploiting the natural gradient at vents. The only energy pathway enzymes present were those of the Wood-Ljungdahl (WL) pathway, which uses H2 as an electron donor and CO2 as electron acceptor. The H2 must have come from geological sources, since it could not have been made through fermentation. Analysis of the phylogenetic trees constructed from the 355 protein families places Clostridia and methanogens as the earliest-diverging organisms - both of which are anaerobic, H2-dependent and use the WL pathway. In methanogens and acetogenic clostridia, methyl groups are central to growth, comprising the very core of carbon and energy metabolism. The implication of this work is that LUCA was very much dependent on abiotic sources of H2 to provide it with energy, consistent with a metabolism associated with lost-city vents in which alkaline mineral rich water enters the acidic high-CO2 ocean.
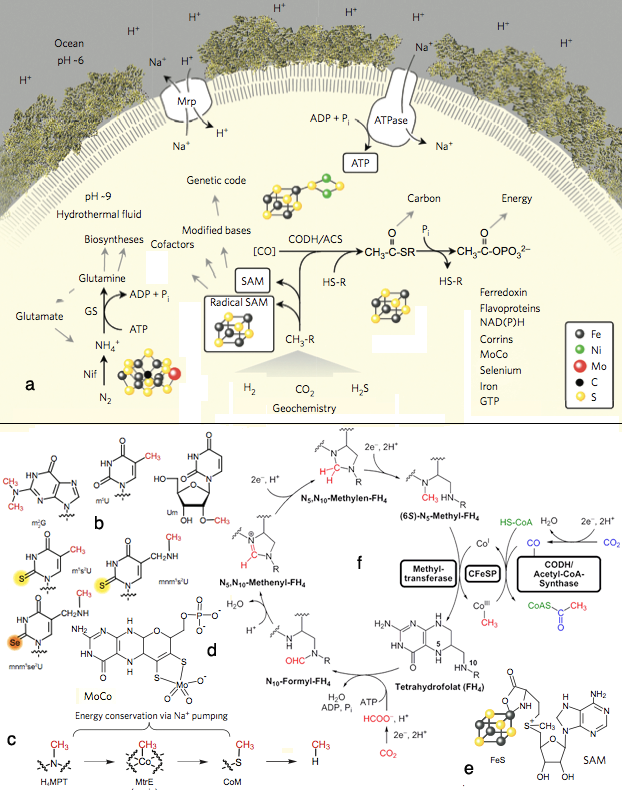 Fig 1c4: Elements of LUCA's metabolism elucidated in Weiss et al. (2016). (a) The overall metabolic pathways iin LUCA. CODH/ACS, carbon monoxide dehydrogenase/acetyl CoA-synthase; Nif, nitrogenase; GS, glutamine synthetase; Mrp, MrP type Na+/H+ antiporter; CH3-R, methyl groups; HS-R, organic thiols. (b) Prominent methyl groups, and S and Se modifications. (c) Methyl transfer from tetrahydrofolate to methane and other C-containing biomolecules. (d) Molybdenum-containg MoCo. (e) SAM S-adenosyl-methionine attached to an FeS center. (f) The WL pathway showing how electron transfer from H2 to CO2 enables incorporation of metabolic molecules.
Fig 1c4: Elements of LUCA's metabolism elucidated in Weiss et al. (2016). (a) The overall metabolic pathways iin LUCA. CODH/ACS, carbon monoxide dehydrogenase/acetyl CoA-synthase; Nif, nitrogenase; GS, glutamine synthetase; Mrp, MrP type Na+/H+ antiporter; CH3-R, methyl groups; HS-R, organic thiols. (b) Prominent methyl groups, and S and Se modifications. (c) Methyl transfer from tetrahydrofolate to methane and other C-containing biomolecules. (d) Molybdenum-containg MoCo. (e) SAM S-adenosyl-methionine attached to an FeS center. (f) The WL pathway showing how electron transfer from H2 to CO2 enables incorporation of metabolic molecules.
Cells conserve energy via chemiosmotic coupling with rotor - stator-type ATP synthases or via substrate-level phosphorylation. LUCA's genes encompass both a phosphotransacetylase (PTA) and an ATP synthase subunit. PTA generates acetylphosphate from acetyl-CoA, conserving the energy in the thioester bond, which can phosphorylate ADP or other substrates. LUCA's WL enzymes are replete with FeS and FeNiS centres, indicating transition-metal requirements and requiring organic cofactors: flavin, F420, methanofuran, two pterins (the molybdenum cofactor MoCo and tetrahydromethanopterin) and corrins such as cobalamin (fig 27), as well as nucleotide and other cofactors.
LUCA's genes for RNA nucleoside modification indicate that it performed chemical modification of nucleosides in both tRNA and rRNA. Four of LUCA's nucleoside modifications are methylations requiring SAM. In the modern code, several base modifications are required for codon-anticodon interactions at the wobble position. Consistent with the recurrent role of methyl groups in LUCA's biology, by far the most common tRNA and rRNA nucleoside modifications that are conserved across the archaeal bacterial divide are methylations, although thio-methylations and incorporation of sulfur and selenium are observed. Notably Selenophosphate synthase is included in the LUCA list, as well as nitrogenase molybdenum-iron protein alpha and beta chains as well as NifH, confirming the LUCA hypothesis for nitrogen fixation (Leigh 2000, Raymond et al. 2004, Boyd & Peters 2013). This picture indicates the antiquity and functional significance of methylated bases in the evolution of the ribosome and the genetic code and forges links between the genetic code, primitive carbon and energy metabolism and hydrothermal environments.
LUCA's gene list reveals only nine nucleotide biosynthesis and five amino acid biosynthesis proteins. The paucity of enzymes for essential amino acid, nucleoside and cofactor biosyntheses suggests that LUCA might not yet have evolved the genes in question prior to the bacterial-archaeal split, with the pathway products for LUCA being still provided by primordial geochemistry.
The late heavy bombardment (LHB) of Earth by comets and asteroids approximately 4-3.8 billion years ago probably resulted in Earth being periodically heated to the point that the oceans were vaporized and probably led to bottlenecks in the diversity of life at the time, meaning that only hyperthermophiles survived. The amount of oxygen available for biological cells was negligible and all life was anaerobic. When we look at the inferred metabolism of LUCA, we are looking at the dominant and most successful kind of metabolism on the planet before the Bacteria and Archaea diverged.
There have been a variety of other studies that have attempted to find a critical minimal gene set for life, or to find the minimal gene set that has homologous members in all three domains. These come to varying conclusions depending on the stringency of their criteria and the choice of representative organisms (Mat et al. 2008, Forterre et al. 2005, Harris et al. 2003).
 To reconstruct the set of proteins LUCA could make, Kim and Caetano-Anollés (2011) (direct link), see also Wang et al. (2007), searched a database of proteins from 420 modern organisms, looking for structures that were common to all. Of the structures found, just 5 to 11 per cent were universal, meaning they were conserved enough to have originated in LUCA. By looking at their function, they conclude that LUCA had an advanced metabolic network, especially rich in nucleotide metabolism enzymes, had primordial pathways for the biosynthesis of membrane glycerol ether and ester lipids, crucial elements of translation, including aminoacyl-tRNA synthases, regulatory factors, and a primordial ribosome with protein synthesis capabilities. It lacked however transcription from DNA to RNA, processes for extracellular communication, and enzymes for deoxyribonucleotide synthesis, and in advanced evolutionary stages stored genetic information in RNA (not DNA) molecules.
To reconstruct the set of proteins LUCA could make, Kim and Caetano-Anollés (2011) (direct link), see also Wang et al. (2007), searched a database of proteins from 420 modern organisms, looking for structures that were common to all. Of the structures found, just 5 to 11 per cent were universal, meaning they were conserved enough to have originated in LUCA. By looking at their function, they conclude that LUCA had an advanced metabolic network, especially rich in nucleotide metabolism enzymes, had primordial pathways for the biosynthesis of membrane glycerol ether and ester lipids, crucial elements of translation, including aminoacyl-tRNA synthases, regulatory factors, and a primordial ribosome with protein synthesis capabilities. It lacked however transcription from DNA to RNA, processes for extracellular communication, and enzymes for deoxyribonucleotide synthesis, and in advanced evolutionary stages stored genetic information in RNA (not DNA) molecules.
Others such as Lane (2005) see the differences between bacterial lipids composed of fatty acids joined to a hydrophilic head by ester bonds and those of archaea utilising cross-linked isoprene molecules joined to the head by ether bonds, with the glycerol phosphate heads having different stereoisomers in archaea and bacteria, as confirming that the membrane arse after the divergence of archaea and bacteria. Lane notes that fermentation did not arise in LUCA as archaea and bacteria use different pathways, confirming the electrochemical basis of the progenitor.
Fig 1d: Phylogenomic tree of proteomes describing the evolution of 420 free-living organisms. phylogenomic study of protein domain structure in the proteomes of 420 free-living fully sequenced organisms. Domains were defined at the highly conserved fold superfamily (FSF) level of structural classification (Kim and Caetano-Anollés).
(click image to link to original research version)
Organelles were thought to be the preserve of eukaryotes, but in 2003 researchers found an organelle called the acidocalcisome also occurred in bacteria. Caetano-Anollés' team has now found that tiny granules in some archaea are also acidocalcisomes, or at least their precursors. That means acidocalcisomes are found in all three domains of life, and date back to LUCA (Seufferheld et al. - direct link).
Acidocalcisomes were originally discovered in Trypanosomes (sleeping sickness and Chagas disease) but have since been found in Toxoplasma gondii (toxoplasmosis), Plasmodium (malaria), Chlamydomonas reinhardtii (a green alga), Dictyostelium discoideum (a slime mould), bacteria and human platelets. Their membranes contain a number of protein pumps and antiporters, including aquaporins, ATPases and Ca2+/H+ and Na+/H+ antiporters. Acidocalcisomes have been implied in osmoregulation. They were detected in vicinity of the contractile vacuole in Trypanosoma cruzi and were shown to fuse with the vacuole when the cells were exposed to osmotic stress. Presumably the acidocalcisomes empty their ion contents into the contractile vacuole, thereby increasing the vacuole's osmolarity. This then causes water from the cytoplasm to enter the vacuole, until the latter gathers a certain amount of water and expels it out of the cell.

Fig 1e: Tangled web linking acidocalcisomes in existent archaea, bacteria and eucaryote species (Seufferheld et al.), overlaying electron micrographs of acidocalcisomes in Agrobacterium tumefaciens(a, b) and Methanosarcina acetivorans (c, d). (click image to link to original research version)
LUCA may have used RNA rather than DNA, as there is no evidence LUCA possessed ribonucleotide reductases, which create the deoxy versions of ribonucleotides the building blocks of DNA (Lundin et al - direct link). Rather it appears these functions have been transferred from bacteria back to archaea by horizontal transfer on at least two separate occasions (arrows in fig 1e). Eucaryotes (mid green) would also have received theirs after LUCA diversification.
 Fig 2: Ribonucleotide reductase trees showing bacterial, eucaryote and archaeal branches, with evidence of two events of horizontal transfer from bacteria to archaea (arrows) after the diversification of LUCA (Lundin et al).
Fig 2: Ribonucleotide reductase trees showing bacterial, eucaryote and archaeal branches, with evidence of two events of horizontal transfer from bacteria to archaea (arrows) after the diversification of LUCA (Lundin et al).
LUCA was a "progenote". Progenotes can make proteins using genes as a template, but the process is so error-prone that the proteins can be quite unlike what the gene specified. Both Di Giulio and Caetano-Anollés have found evidence that systems that make protein synthesis accurate appear long after LUCA. In order to cope, the early cells must have shared their genes and proteins with each other. Caetano-Anollés says the free exchange and lack of competition mean this living primordial ocean essentially functioned as a single mega-organism.
LBCA the last Common Bacterial Ancestor

Fig 2a: Photosynthesis near the Origin of Bacteria
Oliver T et al. (2021) have found that enzymes capable of performing the key process in oxygenic photosynthesis -- splitting water into hydrogen and oxygen -- could actually have been present in some of the earliest bacteria. The earliest evidence for life on Earth is over 3.4 billion years old and some studies have suggested that the earliest life could be older than 4.0 billion years. The team made their discovery by tracing the 'molecular clock' of key photosynthesis proteins responsible for splitting water. They compared the evolution rate of these photosynthesis proteins to that of other key proteins in the evolution of life, including those that form energy storage molecules in the body and those that translate DNA sequences into RNA, which is thought to have originated before the ancestor of all cellular life on Earth. The photosynthesis proteins showed nearly identical patterns of evolution to the oldest enzymes, stretching far back in time.
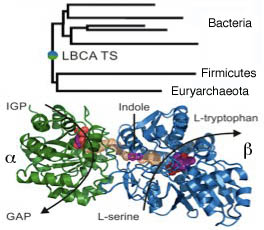 Fig 2b: Ancestral tryptophan synthase (Busch et al.).
Fig 2b: Ancestral tryptophan synthase (Busch et al.).
A picture of the efficiency of enzymes in the last common ancestor of bacteria LBCA, which although more recent than LUCA still dates back some 3.5 byrs, has come from the reverse sequencing of the most probable sequence of the ancestral tryptophan synthase enzyme of the common ancestor of a selection of bacteria and archaea. The tree is rooted within the bacteria, because Euryarchaeota have most likely obtained the TS by a more recent horizontal gene-transfer event from a bacterial predecessor. This proved to have efficient functionality when inserted into E. coli. The LBCA TS subunits are thermostable, exhibit high catalytic activity and form an αββα complex whose crystal structure is similar to modern TS. (Busch et al. 2016).
Some introns, the non-coding sections of DNA that punctuate modular coding sections of eucaryote and some procaryote genes can also self-splice as ribozymes but their link with the RNA era is less encompassing.
Life today is informationally based on the sequences of the four bases A, G, T and C in DNA, with messenger copies of the genetic sequence in mRNA (with U replacing T) forming intermediates in the assembly of proteins, as the cell's primary active chemical and structural agents. This is achieved through a process of translation at the ribosome - a supra-molecular complex composed of some 50 chaperoning proteins surrounding a core composed of three rRNA units, fed by amino-acid coupled tRNAs. The RNAs carry out the essential function, supporting the idea that translation was at first a purely RNA-based process of protein construction. In line with this and other RNA fossils found particularly in Eukaryotes, it is widely believed that life began based on RNA, which shares both the capacity for complementary replication of DNA and the formation of 3-dimensional chemically reactive conformations, similar to proteins, after which the ribosome evolved, transferring the reactive burden on to proteins sequenced through the genetic code. Some time later, the informational genome was consolidated into more stable DNA.
 Fig 3: The initial tree of rRNAs shows three distinct founding domains (Woese 1987) Click to enlarge
Fig 3: The initial tree of rRNAs shows three distinct founding domains (Woese 1987) Click to enlarge
Originally the Bacteria and Archaea were thought to be one large diverse family of prokaryotes until Carl Woese (1977, 1978, 1987, 1990) and others investigated the evolutionary tree of ribosomal RNAs and found that there were three distinct founding evolutionary domains, then named eubacteria, archaebacteria along with the eukaryotes.
This gave the Eukaryotes a closer founding status as well, by contrast with the idea that the procaryotic bacteria came first and then, somehow the higher Eukaryote organisms with their complex cellular structures, including among others - the endoplasmic reticulum, along with the nuclear envelope and Golgi apparatus - all parts of a common complex of internal membranous partitions - and the architecture of microtubules, including centrioles, and the Eukaryote flagellum, as well as the Eukaryotes endosymbiont mitochondria and chloroplasts.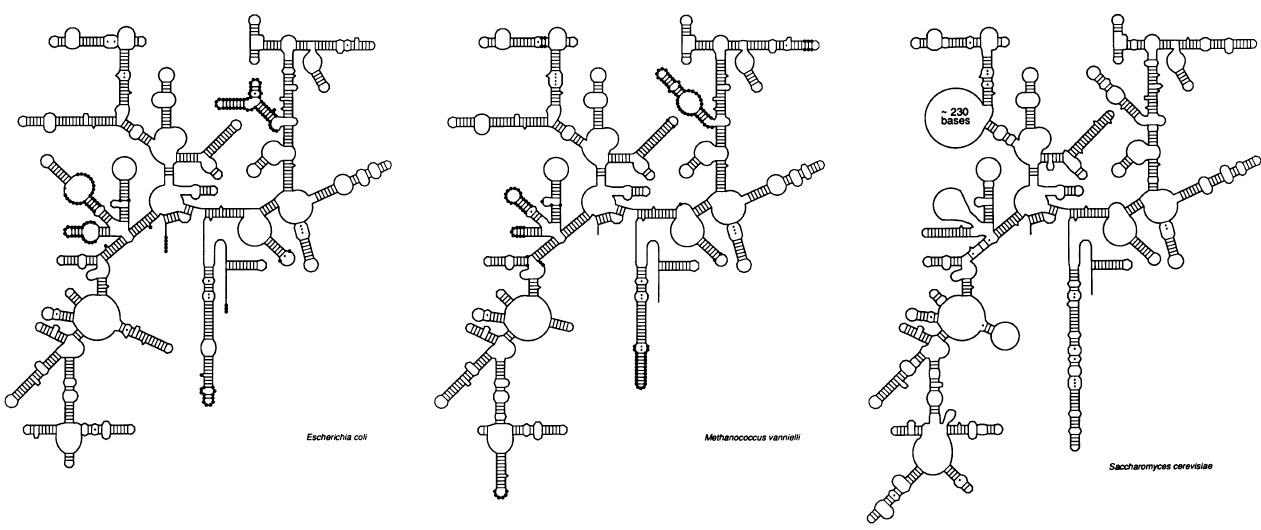
Fig 4: Key structural differences separating
the larger rRNA units of the three domains
(Woese 1987) (click to enlarge).
In addition to their evolutionary sequence divergence, the smaller 30s ribosomal RNAs of each domain, show distinct structural features characteristic of their own domain, but also emphasizing structural links between Bacteria and Archaea on the one hand and Archaea and Eukaryotes on the other, qualitatively confirming the central place of the Archaea in the divergence.


Fig 5: (a) Further elaboration of the rRNA tree (Pace 1997) (b) A third rRNA tree which suggests Archaea lie very close to the root is contrasted with that for the enzyme HMGCoA reductase (c), which also shows evidence of horizontal transfer to an Archaean (ex Doolittle 2000).Right: Revised rRNA tree giving closer correspondence to the archaea-eucarya relationship (Fournier et al. 2010). Click to enlarge
Norman Pace subsequently enlarged the scope and accuracy of the rRNA tree, including a greater diversity of organisms. This tree has become the basis of several other studies. Surviving Archaea are known to inhabit extreme environments, including hot vocanic pools, hydrothermal vents and extreme salty environments and several arrangements of the root of the tree, including Bork's team's work suggest a hot origin for life. However other research (Brochier and Philippe, Boussau et. al.), concludes the base root may have been at about 25oC, a more viable temperature for a simple RNA metabolism, with a succeeding period of high temperature adaptions shortly after the differentiation of the three domains in evolutionary time. A more recent rooting of the ribosomal RNA tree has been produced by Fournier et al. (2010), which coincides more closely with the relationship of archaea and eucarya as sister groups.
However James Lake (1988) had already challenged the notion of three domains, with an analysis claiming that the eucaryotes instead branched off from one line of the archaea, the eocytes or chrenarcheota. This view has been confirmed by accumulating genetic studies (Williams & Embley 2014, Williams et al. 2013, Foster, Cox & Embley 2009, Cox et al. 2008) in which the TACK group of archaeota (fig 5b right) have a pivotal relationship with eucaryotes.


Fig 5b: Left: Evolutionary root of the tree of life and its diversification into archaea, bacteria and Eukaryotes appears to have gone through an early period of cool temperature consistent with an RNA era, followed by a hot period (Anathaswamy, Boussau et. al.) (click to enlarge). Right: Three domains (a) is contrasted with a recent version of the "eocyte" hypothesis (b) showing the eucaryotes emerging from the wider crenarcheota grouping (TACK) after divergence from euryarcheota, implying the amoeboid ancestor of the eucaryotes was an "eocyte" (Williams et al. 2013). Consistent with a eucaryote origin from archaea is the discovery that the DNA in archaea is wrapped around histones in the same manner as eucaryotes, which helps explain how their DNA can survive extremes such as high operating temperatures (Mattiroli et al. 2017), with methanogen histones showing evolutionary correspondence to eucaryote histones.
Virus World: Are Viruses a Complementry Domain of Life?
It is clear that viruses and related elements have coexisted with cellular organisms as far back as the origin of genetic replication. The two form complementary kingdoms the one encapsulated inside a cellular envelope and the other often encased in a polyhedral (generally isocohedral) capsid and depending on cellular metabolisms and replication machinery for predation and/or parasitism of cellular organisms. The transposability of viral genomes also makes them key instruments for 'sexual' mutational transformation of cellular genomes in which they are responsible for the vast amount of horizontal transfer of genetic information, particularly at the tangled roots of the tree of life where a progenote is believed to have consisted of a population fo replicating elements in a mix of cooperative and competitive relationships. Thus a classification of life really consists of two fundamental domains cellular and viral.

Fig 5c: Left: Viral world intermediates between RNA progenote and LUCA( Villarreal L & Witzany G(2009)The two known types of viroid above causing potato spindle disease and replicating asymmetrically in the nucleus and below causing avocado diesase and replicating symmetrically in the chloroplast also functioning as ribozymes in their replication cycle (Daros et al 2006).
At the absolute limit of molecular simplicity that has hints of the RNA era that preceded LUCA are the two genuses of viroid, small circular RNA molecules that cause disease in higher plants. These do not encode any genes, as viruses and all other life forms do, and survive passively through being copied by RNA polymerases, either in the nulceus, or the chloroplasts, using a rolling circle replication that requires no primers or tags. They have pathological effects because their nucleotide sequences cause RNAi interference to essential plant genes, giving rise to popato spindle tuber disease and similar diseases in avocados and other plants. Although they havent been isolated in the wild from cyanobacteria or other prokayotes, they have been found to be able to replicate in cyanobacteria, hinting at an early origin.
Diener's (1989, 2016) hypothesis proposed that the unique properties of viroids make them plausible "living relics" of the RNA world, possessing six primal attributes:
1. viroids' small size, imposed by error-prone replication
2. their high guanine and cytosine content, which increases stability and replication fidelity
3. their circular structure, which assures complete replication without genomic tags
4. existence of structural periodicity, which permits modular assembly into enlarged genomes
5. their lack of protein-coding ability, consistent with a ribosome-free habitat
6. replication mediated in some by ribozymes - the fingerprint of the RNA world.

Fig 5c: Left: Schematic phylogeny of the RNA-dependent RNA polymerase of positive-strand RNA viruses and their capsidless derivatives. The orange lines denote capsidless RNA replicons. Abbreviations: Fu, fungi; Pl, plants; Oo, oomycetes; BDRM, Bryopsis cinicola dsRNA replicon from mitochondria; BDRC, Bryopsis cinicola dsRNA replicon from chloroplasts; SsRV-L, Sclerotinia sclerotiorum RNA virus L. Right: Schematic phylogeny of the RTs of retroelements and the derivative retroviruses. Four major groups of prokaryotic retroelements (gray oval), as well as eukaryotic retroelements and related viruses (blue ovals), are shown. Orange branches represent capsidless retroelements, whereas black branches represent retroviruses, pararetroviruses, and virus-like noninfectious retrotransposons (Metaviridae and Pseudoviridae; dashed black lines). The two large categories of the retroelements are the extrachromosomally primed ones (EP or LTR) and target-primed ones (TP or non-LTR) (Koonin and Dolja 2014).
Moreover although viral genomes are harder to place in a single evolutionary tree due to extensive horizontal transfer of genes, including transfers between the three domains of cellular life as well as among viral elements, research is beginning to elucidate an evolutionary history of the extant viruses that parallels the evolutionary tree of cellular life, leading to the concept of Virusworld (Koonin and Dolja 2014). This world spand a fundamental variety of replications types from single and double-stranded RNA viruses, through retroviruses which use reverse transcriptase to generate a double stranded DNA cope from RNA through to single and double-stranded DNA viruses Many of these form capsids, or spread and enter cells by membrane budding and they also include genetic elements that reside within cells such as DNA transposons, retroelements, LTR elements and antibioti resistance and sexual conjugation pasmidsin bacteria as well as syringe-like bacteriophages Mny of these viral grouping can be traced back to the earliest phases of genetic evolution and so have coexisted with evolving cellular genomes upon which cellular life also despite resisting viral infection has depended on for advantageous mutational innovation.
 Fig 6 Left: Bacterium Gemmata obscuriglobus with internal nuclear envelope and vaccuoles (Rachel Melwig & Christine Panagiotidis / EMBL). Right: Ultrathin EM section of a mimivirus in amoeba (Jean-Michel Claverie) Inset: Mamavirus infected by sputnik phage.
Fig 6 Left: Bacterium Gemmata obscuriglobus with internal nuclear envelope and vaccuoles (Rachel Melwig & Christine Panagiotidis / EMBL). Right: Ultrathin EM section of a mimivirus in amoeba (Jean-Michel Claverie) Inset: Mamavirus infected by sputnik phage.
Offset against both the uniqueness of the mitochondrial endo-symbiosis and the closely linked, but independent question of the origin of the nucleus and nuclear envelope, has been the discovery of giant mimi-, mama-, mega- and pandora-viruses infecting amoeba (Raoult et, al., Philippe et al) and related very large aquatic viruses such as CroV infecting single celled plankton species (Fisher et. al.), which despite their recent discovery, appear from ocean gene analyses to be potentially ubiquitous and widespread in the oceans and possibly playing a crucial role in regulating the atmospheric-oceanic pathways, such as carbon sequestration.
These form an intermediate genetic position between viruses and cells, having the largest genomes, with extensive cellular machinery, including protein translation, and larger than the smallest completely autonomous bacterial and archaeal genomes.
Megavirus chilensis, for example is 10 to 20 times wider than the average virus. The particle measures about 0.7 micrometres (thousandths of a millimetre) in diameter. It just beats the previous record holder, Mimivirus, which was found in a water cooling tower in the UK in 1992. A study of the megavirus's DNA shows it to have more than a thousand genes. The mimivirus genome is a linear, double-stranded molecule of DNA with 1.18 Mbp in length. Megavirus has 1.25 Mbp. Like Mimivirus, Megavirus has hair-like structures, or fibrils, on the exterior of its shell, or capsid, that probably attract unsuspecting amoebas looking to prey on bacteria displaying similar features. These viruses show many characteristics at the boundary of living and non-living. They are as large as several bacterial species, such as Rickettsia conorii and Tropheryma whipplei, possess a genome of comparable size to several bacteria, including those above, and code for products previously not thought to be encoded by viruses. Mimivirus has genes coding for nucleotide and amino acid synthesis, which even some small obligate intracellular bacteria lack. However, it lacks genes for ribosomal proteins, making it dependent on a host cell for protein translation and energy metabolism.
As of mid-2013, an even larger virus with a 2.5 Mb genome without morphological or genomic resemblance to any previously defined virus families has been discovered by the same researchers that found mimivirus, in both the same ocean sample off Peru and in a freshwater pond in Australia. Named pandoravirus - reflecting their lack of similarity with previously described microorganisms and the surprises expected from their future study. The researchers suspect that giant viruses evolved from cells. They think that at some point, the dynasty on Earth was much bigger than the three domains of bacteria, archaea and eukaryotes. Some cells gave rise to modern life, and others survived by parasitizing them and evolving into viruses. Pandora might thus provide a complementary relic of the genomes of this wider founding group (Philippe et al). Using the Global Ocean Sampling (GOS) Expedition data to explore variants of recA (the universal DNA repair enzyme) and rpoB (the beta subunit of bacterial RNA polymerase) a team associated with Craig Venter have discovered branches which may also point to a fourth domain (Wu et al).

Fig 6c: Evolutionary tree of B-family DNA polymerase showing relationship of pandoravirus to other viruses and eucaryotes. Inset is shown pandoraviruses invading acanthamoeba (Philippe et al).
As an illustration of genes in mimivirus normally appearing only in cellular genomes, the mimivirus has genes for central protein-translation components, including four amino-acyl transfer RNA synthetases, peptide release factor 1, translation elongation factor EF-TU, and translation initiation factor 1. The genome also exhibits six tRNAs. Other notable features include the presence of both type I and type II topoisomerases, components of all DNA repair pathways, although the topoisomerase 1B has a different header structure from the eucaryote form (Brochier-Armanet, Gribaldo & Forterre 2008), many polysaccharide synthesis enzymes, and one intein-containing gene. Inteins are protein-splicing domains encoded by mobile intervening sequences (IVSs). They self-catalyze their excision from the host protein, ligating their former flanks by a peptide bond. They have been found in all domains of life (Eukaria, Archaea, and Eubacteria), but their distribution is highly sporadic. Only a few instances of viral inteins have been described. Self-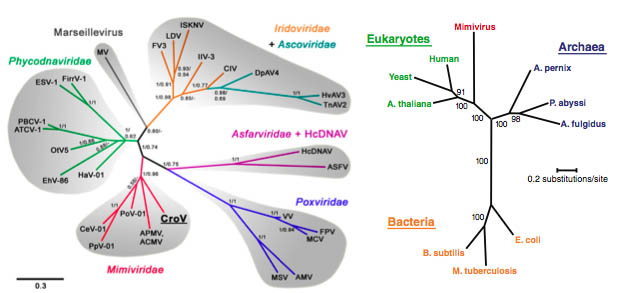 splicing type I introns are a different type of mobile IVS, self-excising at the mRNA level. They are rare in viruses. Mimivirus exhibits four instances of self-excising intron, all in RNA polymerase genes.
splicing type I introns are a different type of mobile IVS, self-excising at the mRNA level. They are rare in viruses. Mimivirus exhibits four instances of self-excising intron, all in RNA polymerase genes.
Fig 6d: Evolutionary diversification of Mimiviruses from nucleocytoplasmic large DNA viruses (Fisher et. al.) and in relation to the three domains of cellular life based on the concatenated sequences of seven universally conserved protein sequences (Raoult et. al.)
Mamaviruses also host parasitic virophages, affectionately named sputnik (Pearson 2008) as viral satellites, which piggy back on the metabolism of the large viral factories set up by these giant viral genomes causing the mimiviruses to sicken, and these virophages also contains genes that are linked to viruses infecting each of the three domains of life Eukarya, Archaea and Bacteria (La Scola et. al.). It has thus been suggested that they have a primary role in the establishment of cellular life and that they may have been instrumental in the emergence of the nuclear envelope
It as been suggested the the even larger klosneuviruses, with genome sizes up to 3 Mb (Schultz et al. 2017) shows they have arisen through multiple aggregation events, but researchers studying the tupanviruses (Abrahão et al. 2018) with 1.5 Mb genomes and a wide array of amino acid genes suggest they may have arisen from older giant viruses by genome reduction as obligate parasites.
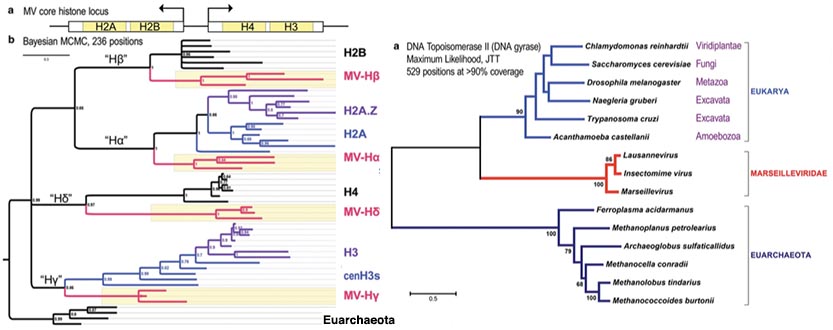
Fig 6e: Evolutionary relationships between histone complexes and topoisomerase II of marseilleviruses places their incorporation as to or from an ancestor of LECA
before eucharyote histone structure had fully evolved (Erives 2017).
Yet another group of nucleocytoplasmic large DNA viruses (NCLDV) of eukaryotes, are typified by Marseille virus (Boyer et al. 2009) a giant virus of amoeba, prototypical of the family Marseilleviridae (MV) has been found to harbor core histone doublets consistent with incorporation from an ancient precursor of LECA the last common ancestor of eucaryotes. The genome of the virus is composed of typical NCLDV core genes and genes apparently obtained from eukaryotic hosts and their parasites or symbionts, both bacterial and viral. The virions of Marseillevirus encompass a 368-kb genome, a minimum of 49 proteins, and some messenger RNAs. The genetic sequences of the histone doublets places them at the root of the Eucaryote tree and DNA topoisomerases also present in the virus are likewise consistent with an origin close to the divergence of euarchaeota and eucaryotes (Erives 2017).
 Fig 6f: Evolutionary tree of protein-primed B family DNA polymerases leading to casposons, including a variety of viruses and prokaryotic and eucaryotic plasmids.
Fig 6f: Evolutionary tree of protein-primed B family DNA polymerases leading to casposons, including a variety of viruses and prokaryotic and eucaryotic plasmids.
CRISPR-Cas has become famous for its potential to perform genome editing. About 90% of archaea and 30% of bacteria have some form of CRISPR-Cas immunity, but how did bacteria and archaea come to possess such sophisticated immune systems? Viruses outnumber prokaryotes by ten to one and are said to kill half of the world's bacteria every two days. Prokaryotes also swap scraps of DNA called plasmids, which can be parasitic. Prokaryotes have evolved a slew of weapons to cope with these threats. Restriction enzymes, for example, cut DNA at or near a specific sequence, but these defences are blunt. Each enzyme is programmed to recognize certain sequences, and a microbe is protected only if it has a copy of the right gene. CRISPR–Cas is more dynamic. It adapts to and remembers specific genetic invaders in a similar way to how human antibodies provide long-term immunity after an infection. The leading theory of their origin is that the systems are derived from transposons that can hop from one position to another in the genome. Evolutionary biologist Eugene Koonin and colleagues Krupovic et al. (2014) have found a class of these they have called Casposons that encode the protein Cas1 involved in inserting spacers into the genome. They describe a new superfamily of archaeal and bacterial mobile elements which encode Cas1 endonuclease, a key enzyme of the CRISPR-Cas adaptive immunity systems of archaea and bacteria. The casposons share several features with self-synthesizing eukaryotic DNA transposons of the Polinton/Maverick class, including terminal inverted repeats and genes for B family DNA polymerases. However, unlike any other known mobile elements, the casposons are predicted to rely on Cas1 for integration and excision, via a mechanism similar to the integration of new spacers into CRISPR loci.
Another very ancient virus defence and RNA processing enzyme also related to some CRISPR systems, spans all the domains of life. The RNase III enzyme Drosha is the core nuclease that executes the initiation step of microRNA (miRNA) processing in the eucaryote nucleus, further processed by related Dicer. The microRNAs thus generated are short RNA molecules that regulate a wide variety of other genes by interacting with the RNA-induced silencing complex (RISC) to induce cleavage of complementary messenger RNA (mRNA) as part of the RNA interference pathway. RNAase III is essential in prokaryotes to slice up rRNA precursors into ribosomal RNAs and appear to function based on the stem-loop structure of the substrate, rather than specific sequences. Drosha and the family of RNAse III enzymes it now emerges were likely to have also been original virus fighters in a single-celled ancestor of animals and plants. Some CRISPR systems in archaea and bacteria also include RNAse III proteins suggesting they have an evolutionary relationship or that some CRISPR systems adopted an existing RNAase III activity. Eucaryotes are likely to have retained this ancient system because their active use of RNA has also left them uniquely exposed to RNA viral attack. Plants and invertebrates deploy RNAse III proteins in an immune response called RNA interference, or RNAi, slicing the invader's RNA into chunks that prevent it from spreading. Vertebrates ward off viruses with interferons — while Drosha and a related protein regulate genes in the nucleus. However vertebrate Drosha has also been found to tackle viruses (Aguardo 2017).
Tangled Roots of Horizontal Transfer
Darwin (1859) was the first person to publish an evolutionary tree of life on page 133 of his seminal work. The basis of such a tree in the genetic age has become the vertical transfer of genetic information through reproduction coupled to mutation and selective advantage. This is the basis of the tree diagram itself and all evolutionary trees constructed on genetic data. However, despite the division into three domains, further investigations of proteins in the three domains began to reveal a much more confused and complicated picture.
 Fig 7: Tangled roots (Doolittle 2000)
Fig 7: Tangled roots (Doolittle 2000)
Firstly the ribosomal proteins, like the rRNAs show distinct, easily differentiated morphologies with some correspondences linking one pair of domains and other another pair (Forterre 2006b, Woese 2000). Secondly, horizontal transfer of genes e.g. through viral interaction has occurred at fluctuating rates throughout all the domains of life. Lawton (2009), provides an in depth a review of this debate. Thirdly, the proteins in Eukaryotes appear to have a mixed origin, with the informational ones having an evolutionary relationship with archaea but the metabolic enzymes appearing to have a bacterial origin. This suggests that the Eukaryote genome has either resulted from one, or more symbiotic fusions e.g. an archaeal and a bacterial genome and/or that there has been a high degree of horizontal gene transfer between bacteria and Eukaryotes.
The evidence for symbiotic inclusions is clear from the fact that all Eukaryotes have endosymbiotic respiring mitochondria. Plants also have photosynthetic chloroplasts derived from cyanobacteria. The only apparent exceptions are a few primitive anaerobic organisms, such as the metamonad human gut parasite Giardia lamblia which nevertheless has a mitochondrial remnant.
There are glaring incidences of horizontal transfer in higher organisms, where for example, cows have a gene that originated in snakes. The picture of horizontal transfer is even more tangled in bacterial and archaeal genomes, which contain a great number of shared and exchanged genes, through promiscuous viral transfer between species.
This has led to Doolittle (1998), Woese (2002), and others, proposing a tangled root to the tree of life involving a transition from a regime in which there was a much higher rate of horizontal exchange and effective global optimization of genomes, to tree-like vertical evolution of genomes, once the more complex genomes of the Eukaryote domains became established.

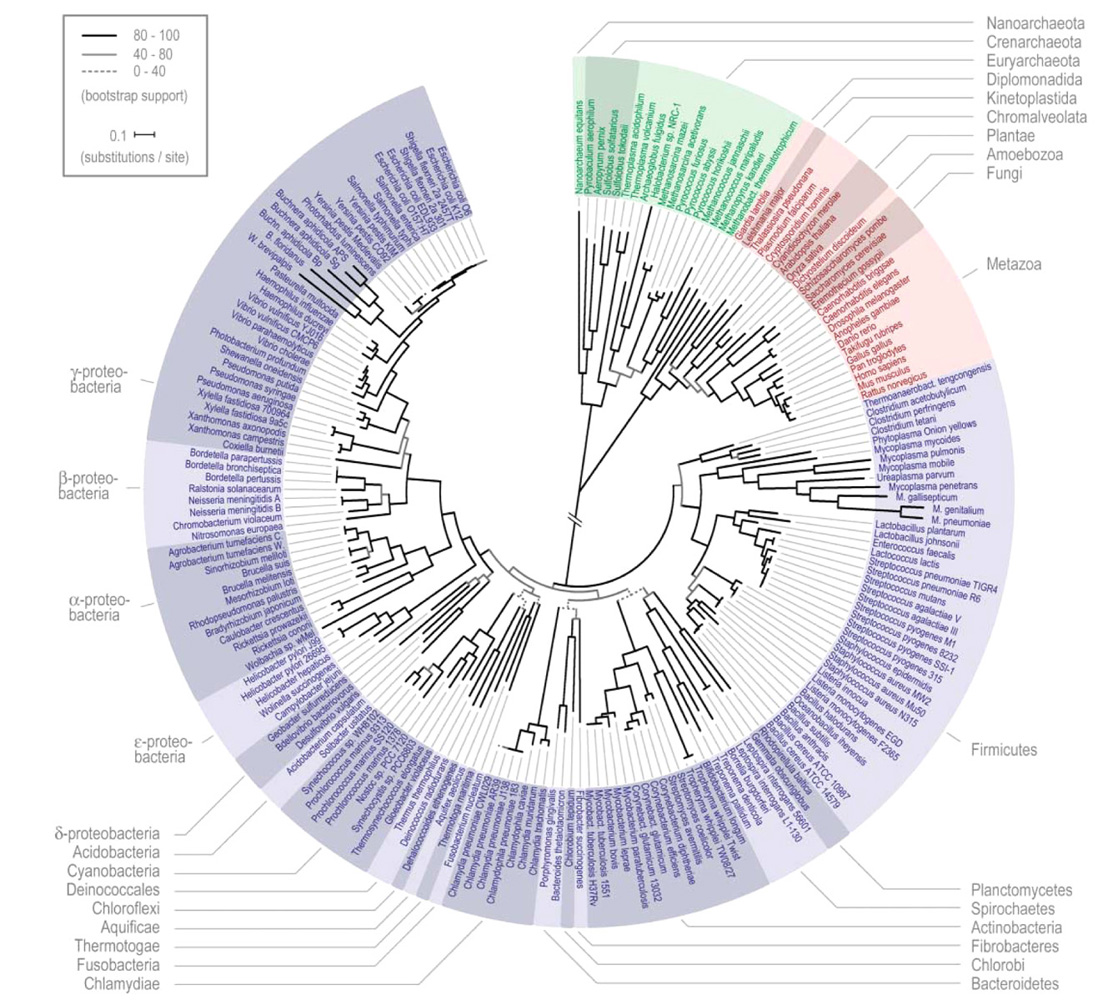
Fig 8,9: Left: Superfamily fold incidence evoutionary tree of eucaryotes and the three domains of life. The number of folds connecting each group is shown lower left (Yang, Doolittle & Bourne 2005).
Right: High resolution tree of the three domains of life - eubacteria, archaea, eukaryotes. (Ciccarelli, Bork et. al. 2006) (click to enlarge).
One way of testing whether the three branches actually have a meaningful evolutionary tree is to use the simple presence of a given super-family protein fold as a classifier (Yang, Doolittle & Bourne 2005). This proves to be a more accurate measure of taxonomy than many others, based of fold frequency, which correctly divides the Archaea into crenarchs and euryarchs and groups the Eukarya into animals, plants, fungi, and others (protists). This method leads to the evolutionary tree illustrated left in fig 9. This leads to the implication that LECA was a well-defined organism with a rich gene complement and not just a quasi-genome shaped by horizontal transfer.
To try to clarify the taxonomic relationships founding the tree of life, Peer Bork and his team produced a refined evolutionary tree (fig 9 right) by selecting only universal proteins that had not been subjected to horizontal transfer, providing the most detailed tree root diagram to date, although admittedly on only a skeleton gene set comprising some 1% of the respective genomes. The phylogenetic tree has its basis in a cleaned and concatenated alignment of 31 universal protein families and covers 191 species whose genomes have been fully sequenced. Merhej et. al. have further demonstrated convergent evolution among specialized bacterial groups.
 Fig 10: Tree diagram of the birth, transfer, duplication and loss of key genes in the redox and electron transport pathways, in a founding burst of gene evolution between 3.3 and 2.7 billion years ago (David and Alm 2010).
Fig 10: Tree diagram of the birth, transfer, duplication and loss of key genes in the redox and electron transport pathways, in a founding burst of gene evolution between 3.3 and 2.7 billion years ago (David and Alm 2010).
Then Lawrence David and Eric Alm (2010) produced the above tree investigating the central genes common to a wide spectrum of life forms, involving the founding steps of redox reactions and electron transport, demonstrating a rapid evolutionary innovation during an Archaean genetic expansion between 3.3 and 2.7 billion years ago. They mapped the evolutionary history of 3983 gene families that occur in a wide range of modern species. They were able to show that 27 per cent of these gene families appeared in a short evolutionary burst. Many of the genes from this time were involved in electron transport - a key step in respiration and photosynthesis, which ultimately led to oxygen-producing photosynthesis and the "great oxygenation event" 2.4 billion years ago, when the atmosphere became oxygen rich.
The Great Oxygenation Event and the Origin of Oxygen Photosynthesis
This lends support to the idea that the collective primordial genome functioned as a supercomputer (King 2010) based on parallel genetic algorithms combined with horizontal genetic transfer, whose bit computation rate through mutation and recombination is sufficient to generate the functional conformations, through protein folding, to solve the key metabolic pathways over a period no longer than 300 million years.
 Fig 10b: Tree of orphan genes in metazoa with charts for the mouse and fruit fly, show the emergence of orphans throughout the span of evolution, with a peak in both at 800 million years ago when earth emerged from its “snowball” phase, with the current peaks corresponding to newborn genes, many of which will be lost. About 20 percent of new genes in fruit flies appear to be required for survival. And many others show signs of natural selection (Tautz & Domazet-Loso 2011).
Fig 10b: Tree of orphan genes in metazoa with charts for the mouse and fruit fly, show the emergence of orphans throughout the span of evolution, with a peak in both at 800 million years ago when earth emerged from its “snowball” phase, with the current peaks corresponding to newborn genes, many of which will be lost. About 20 percent of new genes in fruit flies appear to be required for survival. And many others show signs of natural selection (Tautz & Domazet-Loso 2011).
Contrasting with the the early emergence of key functional genes it has been discovered that regions of non-coding DNA have been repeatedly activated to become de-novo 'orphan' genes, which cannot be accounted for by gene duplication, conversion to new functions through exon shuffling to make new modular arrangements, or genes generated from transposable elements. While orphan genes might seem exponentially improbable on the basis that n base pairs with 4n possible arrangements could randomly become functional, such genes have been recently found to be ubiquitous. 10-20% of genes in all taxa so far explored lack homologs in other species. About 2/3 of domains of unknown function (DUF) open reading frames (ORF) whose 3-D structures have been analysed show folds which are likely outlier extremes of existing gene families not recognized by gene comparison systems such as BLAST (Jaroszewski et al. 2009) many are also de-novo orphans.
A clear example is the Pldi gene in the mouse M. musculus, which has arisen within the past 2.5-3.5 million years in a large intergenic region present in many mammals, including humans, thus excluding gene duplication, transposable elements, or other genome rearrangements. The gene has three exons, shows alternative splicing, and is specifically expressed in postmeiotic cells of the testis enhancing sperm motility (Heinen et al. 2009). Its emergence correlates with indel mutations in the 5' regulatory region. A recent selective sweep is associated with the transcript region in M. musculus populations.
In humans, at least one de novo gene is active in the brain, leading some scientists to speculate such genes may have helped drive the brain’s evolution. Others are linked to cancer when mutated, suggesting they have an important function in the cell. De novo genes are often short, and produce small proteins. Rather than folding into a precise structure they have a more disordered architecture, allowing the protein to promiscuously bind to a broader array of molecules (Singer 2015). Investigations suggest that such taxon-specific genes drive morphological specification, enabling organisms to adapt to changing conditions in the generation of morphological diversity, and innate defence (Khalturin et al. 2009).

Fig 10c: Archaeal and Bacterial Trees (Parks et al. (2017) identified by metagenomic genome assemblyfromdiverse fragments collected in the wild
extending known phylla (green) with 30% more species and discovering new phylla (red).
Bacteria and Archaea engage in much more radical forms of pan-sexuality than higher organisms, involving viruses and plasmids, themselves separate mobile genetic elements, acting as agents of genetic transfer, accelerating the pace of bacterial evolution (Maxmen 2010). This enables the genetic sequences of bacteria, archaea and protists to move around in the genome and to be exchanged between cells, and even between different species. Sexual exchange of material can happen both through viral exchange and through a conjugation plasmid, which can spool DNA from one bacterium into another, resulting in a net donation of genes from one strain or species to another, which ensures a broad exchange of genetic material throughout bacterial ecosystems, resulting in rapid accumulation of advantageous genes exemplified by plasmid borne infectious drug resistance.
To give a very rough idea of the computing power of the combined bacterial genome alone, taking into account bacterial soil densities (~109/g), effective surface area (~1018 cm2), genome sizes (~106), combined reproduction and mutation rates (~10-3/s) gives a combined presentation rate of new combinations of up to 1030 bits per second, roughly 1012 times greater than the current fastest computer at 33 petaflops or about 1017 bit ops per second. Corresponding rates for complex life forms would be much lower, at around 1017 per second because they are fewer in total number and have lower reproduction rates and longer generation times, but they are still vying with the computation rates of the fastest supercomputer on earth.

An even higher figure has been given by Landenmark et al. (2015). Using information on the typical mass per cell for each domain and group and the genome size, estimate the total amount of DNA in the biosphere to be 5.3 x 1031 (±3.6 x 1031) megabases (Mb) of DNA (Table 1). This quantity corresponds to approximately 5 x 1010 tonnes of DNA, assuming that 978 Mb of DNA is equivalent to one picogram. Assuming the commonly used density for DNA of 1.7 g/cm3, then this DNA is equivalent to the volume of approximately 1 billion standard (6.1 x 2.44 x 2.44 m) shipping containers. The DNA is incorporated within approximately 2 x 1012 tonnes of biomass and approximately 5 x 1030 living cells, the latter dominated by prokaryotes. By analogy, it would require 1021 computers with the mean storage capacity of the world’s four most powerful supercomputers (Tianhe-2, Titan, Sequoia, and K computer) to store this information. If all the DNA in the biosphere was being transcribed at reported rates, taking an estimated transcription rate of 30 bases per second, then the potential computational power of the biosphere would be approximately 1015 yottaNOPS (yotta = 1024), about 1022 times more processing power than the Tianhe-2 supercomputer, which has a processing power of 33.86 peta flops on the order of 105 teraFLOPS (tera = 1012).
This picture of bit rates coincides closely with the Archaean expansion scenario noted above and suggests that evolution has been a two-phase process in  which the much higher bit rates of the collective single-celled genome under promiscuous sexuality and horizontal transfer has arrived at a global genetic solution to the protein folding problems of the central metabolic, electro-chemical and even root developmental pathways, which are then later capitalized on by multi-celled organisms, through gene duplication and loss as well as the creation of new specialized genes at a much lower rate.
which the much higher bit rates of the collective single-celled genome under promiscuous sexuality and horizontal transfer has arrived at a global genetic solution to the protein folding problems of the central metabolic, electro-chemical and even root developmental pathways, which are then later capitalized on by multi-celled organisms, through gene duplication and loss as well as the creation of new specialized genes at a much lower rate.
Fig 11: Horizontal transfers across the bacterial tree under two thresholds 10, 5 genes (Dagan et. al.).
The massive extent of horizontal transfer in eubacteria, as well as archaea has also become clear suggesting large components of procaryote genomes are effectively globaly optimized for their niches by frequent genetic transfer. Dagan et. al. have characterized the extent of horizontal transfer for a series of thresholds as well as establishing specific modularity of horizontal transfer of functions between groups.
 Fig 12: Genetic diffusion at the root of the tree (Dagan and Martin)
Fig 12: Genetic diffusion at the root of the tree (Dagan and Martin)
Critics of the validity of the tree root concept, such as Dagan and Martin emphasize the small proportion (1%) of the genome used in Bork's study and stress both the lateral (or horizontal) gene transfer events uniting the prokaryote realms and the apparent chimaeric nature of the Eukaryote genome, which appears to contain both archaea-related informational genes and eubacterial metabolic ones, in addition to obvious endosymbiont events of the mitochondrion and chloroplast.
The case for horizontal transfer of genes between unrelated Eukaryote species through infectious elements invading new and hence non-resistant species is also well established. Genome-wide comparative and phylogenetic analyses show that HGT in animals typically gives rise to tens or hundreds of active ‘foreign’ genes, largely concerned with metabolism (Crisp et al. 2015). The SPIN element is present in a diverse unrelated set of species, spanning amphibians, reptiles, marsupials and mammals while absent from closely related species (Lisch).
 Fig 13: Left: Spread of the HAS1 hyaluronan synthase genes across diverse groups - chordates, other metazoa, fungi, plants, bacteria and archaea (Crisp et al. 2015). Right: Pattern of invasions of the SPIN element (Lisch)
Fig 13: Left: Spread of the HAS1 hyaluronan synthase genes across diverse groups - chordates, other metazoa, fungi, plants, bacteria and archaea (Crisp et al. 2015). Right: Pattern of invasions of the SPIN element (Lisch)
The estimate of the number of phyla of as yet undiscovered bacteria continues to grow in an unbounded trajectory. A research team from Berkeley (Brown et al. 2015) gathered water samples from the Colorado River, passed the water through a pair of increasingly fine filters - with pores 0.2 and 0.1 microns wide - and then analyzed the bacteria captured by the filters, many of which were very small and included hair-like structures (inset fig 13b), reconstructing the scrambled short pieces of DNA into complete and draft genomes. They divided the 789 organisms into 35 phyla - 28 of which were newly discovered.  They based the sorting on the organisms’ evolutionary history and on similarities in the code on the organisms’ 16S rRNA genes - those with at least 75% of their code in common went into the same phylum. With these new additions, there are now roughly 90 identified bacterial phyla. This is a lot more than there were a year ago, but also far fewer than the 1,300 to 1,500 phyla that microbiologists estimate we’ll have once a complete accounting is finished.
They based the sorting on the organisms’ evolutionary history and on similarities in the code on the organisms’ 16S rRNA genes - those with at least 75% of their code in common went into the same phylum. With these new additions, there are now roughly 90 identified bacterial phyla. This is a lot more than there were a year ago, but also far fewer than the 1,300 to 1,500 phyla that microbiologists estimate we’ll have once a complete accounting is finished.
Fig 13b: New phyla of bacteria (Brown et al. 2015).
The Eukaryote Nuclear Genome as a Genetic Fusion
 Fig 14: The proposed ring of life (Rivera and Lake)
Fig 14: The proposed ring of life (Rivera and Lake)
Rivera and Lake used a new algorithm to take account of possible genetic fusion events, forming a genetic ring through matching partial trees into a most parsimonious whole, inferring that the Eukaryote genome has arisen from a fusion of an archaeal (possibly eocyte) genome with that of either a cyanobacterium or possibly a γ-proteobacterium.
The method used cannot definitively determine whether or not the eubacterial genome could have come from the mitochondrial event, which, to an even greater extent than the more recent chloroplast, has resulted in a high net transfer of genes from the mitochondrial chromosome to the nucleus, leaving open the possibility that in addition to the mitochondrial symbiosis, and the later chloroplast one, there may have been an additional genetic fusion. Lane and Archibald have cited further major endosymbiosis events involving complex three genome interaction in protista, where both green and red algae have been incorporated by endosymbiosis into other protists, which demonstrate both that endosymbiois has occurred many times and the genomic complexity of nuclear symbiont gene exchange.


Fig 15: Left: Separate archaeal and bacterial origins of informational and operational genes (Rivera et al.). Proposed fusion between two genomes - informational, from Archaea (red), and metabolic, from Eubacteria (blue) as well as mitochondrial genes migrating to the nucleus (green) (Horiike et. al.). Up to 75% of nuclear genes whose ancestry has been elucidated may come from bacteria (Lane).
The idea of a genetic fusion between a member of the archaea and a proteobacterium is also supported by several other lines of research. Rivera et al. (1992) had demonstrated genomic evidence for two functionally distinct gene classes. The deeper, diverging informational lineage codes for genes which function in translation, transcription, and replication, and also includes GTPases, vacuolar ATPase homologs, and most tRNA synthetases. The more recently diverging operational lineage codes for amino acid synthesis, the biosynthesis of cofactors, the cell envelope, energy metabolism, intermediary metabolism, fatty acid and phospholipid biosynthesis, nucleotide biosynthesis, and regulatory functions. In eukaryotes, the informational genes are most closely related to those of Methanococcus. Evolution of glycolytic enzymes is consistent with this idea (Emelyanov). ‘Homology-hit analysis’ of non-mitochondrial genes determined the number of yeast orthologous ORFs in each functional category to the ORFs in six Archaea and nine Bacteria at several thresholds, suggesting an archaeal parasite engulfed by a eubacterium (Horiike et. al.) the proposal that close association between a central methanogenic archaebacterium (archaea) and a close-knit surrounding clump of ancestral sulfate-respiring δ-proteobacteria could have also led to the nucleus and endoplasmic reticulum (Moreira and Lopez-Garcia). See also fig 16a2.
Fig 15b: Reduction in the number of mitochondrial protein-coding genes has occurred in several waves of exopnential loss, varying among the principal eukaryote groups
supporting an original division early in the endosymbiosis (Janouskovec et al. 2017).
Recent work shows that the mitochondrial and nuclear genomes are in a feedback relationship, rather than the former being merely a denuded genetic skeleton reduced to its bare bones functions. While the process of transfer of mitochondrial genes to the nucleus has resulted in a tally of mitochondrial genes of only 37 in humans, comprising respiration enzymes and essential genes involved in mitochondrial DNA and protein synthesis, the highly-compactified human 16,569 bp mitochondrial genome nevertheless contains up to 500 overlapping open reading frames (Yen et al. 2013), as well as abundant mitochondrial genome-encoded small RNAs (Ro et al. 2013), which appear to be products of currently unidentified mitochondrial ribonucleases and may have a regulatory role. Humanin, a ubiquitous protein involved in human stress protection (Yen et al. 2013) appears to be generated from the mitochondrial genome, although duplicate copies also appear to have been transferred to the nucleus. This is also  consistent with the metabolically responsive roles of the mitochondrion, for example in in initiating apoptosis (controlled cell death) in which humanin plays an inhibitory role, and enriching synaptic contacts in neurons (Sun et al. 2013). Mitochondrial genomes evolve 5-15 times faster than the nuclear genome. Until recently, it was generally accepted that all mitochondrial DNA molecules are identical at birth, however, recent work has shown that ~25% of healthy individuals inherit a mixture of wild-type and variant mtDNA, generally involving the non-coding hypervariable mtDNA D-loop responsible for initiating DNA replication and transcription (Payne et al. 2013). Variant mtDNA lineage expansions have been found in Tibetan sherpas living at different altitudes suggesting evolutionary adaptions over the order of 102 years (Kang et al. 2013). In turn, it has also been found that endurance exercise can turn over mitochondria, effectively removing those with reduced function due to accumulated mutations (Safdar et al. 2011).
consistent with the metabolically responsive roles of the mitochondrion, for example in in initiating apoptosis (controlled cell death) in which humanin plays an inhibitory role, and enriching synaptic contacts in neurons (Sun et al. 2013). Mitochondrial genomes evolve 5-15 times faster than the nuclear genome. Until recently, it was generally accepted that all mitochondrial DNA molecules are identical at birth, however, recent work has shown that ~25% of healthy individuals inherit a mixture of wild-type and variant mtDNA, generally involving the non-coding hypervariable mtDNA D-loop responsible for initiating DNA replication and transcription (Payne et al. 2013). Variant mtDNA lineage expansions have been found in Tibetan sherpas living at different altitudes suggesting evolutionary adaptions over the order of 102 years (Kang et al. 2013). In turn, it has also been found that endurance exercise can turn over mitochondria, effectively removing those with reduced function due to accumulated mutations (Safdar et al. 2011).
Bacteria generally have a nucleoid region of distinctive cytoplasm around the DNA, but no nuclear membrane. An intriguing question is raised by the bacterium Gemmata obscuriglobus (fig 6) which is a member of the phylum Planctomycetes (see fig 9 enlargement) which appears to have both a nuclear envelope and endoplamic reticulum-like intra-cellular membranes and the ability to uptake proteins present in the external milieu in an energy-dependent process analogous to eukaryotic endocytosis consistent with autogenous evolution of endocytosis and the endomembrane system in an ancestral non-eukaryote cell (Lonhiennea et. al.).
Fig 16: Gene replacement tree root (Makarova) Hartman and Federov's list of putative chronocyte genes correspond to the 359 above.
Other theories stick to the three domain paradigm and propose that a primitive Eukaryote precursor possibly still retaining an RNA-based genome, as suggested by Woese (1998) might be the case for the progenote (first root life form) to be the last universal common ancestor of all three domains, possibly including genes for endoplasmic reticulum and microtubules, which later engulfed both a member of the archaea and a eubacterium. Hartman and Federov cite a collection of such genes, including those for ribosomal proteins as well, naming the organism as a chronocyte.
This is also consistent with the much greater complexity of use of RNA in Eukaryotes, including alternative splicing, the use of introns, interfering-RNAs in gene regulation, micro-RNAs and the use of the nucleus to contain a diversely functioning RNA informational metabolism not unlike that of a putative progenote.
LECA: Finding the Roots of the Eucaryote and Metazoan Trees
The Copernican principle asserts that the Earth is a typical rocky planet in a typical planetary system, located in an unexceptional region of a common barred-spiral galaxy, hence it is probable that the universe teems with complex life. This is supported to a reasonable extent by the discovery of an increasing number of planets including some putative "Goldilocks" zone planets where water would be liquid and life as we know it could potentially exist. Set against this, the "rare earth" hypothesis argues that the emergence of complex life requires a host of fortuitous circumstances, including a galactic habitable zone, a central star and planetary system having the requisite character, the circumstellar habitable zone, the size of the planet, the advantage of a large satellite, conditions needed to assure the planet has a magnetosphere and plate tectonics, the chemistry of the lithosphere, atmosphere, and oceans, the role of "evolutionary pumps" such as massive glaciation and rare bolide impacts, and whatever led to the still mysterious Cambrian explosion of animal phyla. This might mean that planets able to support a bacterial level of life are not so uncommon, but those supporting complex multicellular life might be.
 Fig 16: Metabolic power of eucarote cells per haploid genome and hence the capacity for genomic complexity depends on the respiratory power of mitochondria (Lane and Martin).
Fig 16: Metabolic power of eucarote cells per haploid genome and hence the capacity for genomic complexity depends on the respiratory power of mitochondria (Lane and Martin).
Bringing this question to a pivotal crux in our context, the emergence of mitochondria as endosymbionts has been proposed to be a critical bottleneck which allowed complex life to evolve only once on Earth, because, only in this effectively fractal cellular architecture, can the membrane surface areas necessary to support the chemical reactions enabling the vastly larger number of genes in a complex organism's genome to maintain metabolic stability (Lane and Martin 2010, 2012). Lane and Martin note "The cornerstone of eukaryotic complexity is a vastly expanded repertoire of novel protein folds, protein interactions and regulatory cascades. The eukaryote common ancestor increased its genetic repertoire by some 3,000 novel gene families. The invention of new protein folds in the eukaryotes was the most intense phase of gene invention since the origin of life. Eukaryotes invented five times as many protein folds as eubacteria, and ten times as many as archaea. Even median protein length is 30% greater in eukaryotes than in prokaryotes". Whether such endo-symbiosis is rare. or a common extreme of parasitic or predatory relationships would then determine how likely or unlikely complex life might be.
There are three ideas of how mitochondrial endosymbiosis could have occurred.
Fi g 16a: Evolution of iron-sulphur cluster proteins of mitochondria is linked to α-proetobacteria (Emelyanov 2003)
g 16a: Evolution of iron-sulphur cluster proteins of mitochondria is linked to α-proetobacteria (Emelyanov 2003)
The first theory, which places the emphasis on archaea phagocytosis, consists of an archaeal cell e.g. from the TACK group (figs 5b, 16a) engulfing an α-proteobacterium similar to Rickettsia, which has known homology with mitochondria (Andersson 1998). Mitochondria are evolutionarily related to α-proteobacteria and in particular the SAR11-clade of Rickettsiiae (Emelyanov 2003, Thrash et al. 2011) named after their discovery in the Sargasso sea. SAR11 clade organisms, unlikeRickettsiae prowazekii responsible for typhus, are free living dominant ocean organisms. Pelagibacter ubique discovered in the Sargasso sea, along with its relatives constitute 25% of all microbial plankton cells - the most abundant ocean bacteria and possibly the most abundant bacteria on Earth. It is one of the smallest self-replicating cells known, with a length of 0.37-0.89 µm and a diameter of only 0.12-0.20 µm. 30% of the cell's volume is taken up by its genome. It has the smallest genome 1.30 Mbp of any free living organism, encoding only 1,354 open reading frames (1,389 genes total). The only species with smaller genomes are intracellular symbionts and parasites, such as Mycoplasma genitalium. It recycles dissolved organic carbon and undergoes regular seasonal cycles in abundance - in summer reaching ~50% of the cells in the temperate ocean surface waters. Thus it plays a major role in the Earth's carbon cycle.
 Fig 16a2: Nitrosopumilus maritimus one of the ubiquitous Thaumarchaea
Fig 16a2: Nitrosopumilus maritimus one of the ubiquitous Thaumarchaea
and Pelagibacter ubique from the SAR11 clade.
Complementing this, in terms of close archaeal relatives of the eucaryote endosymbiosis, are the Crenarchaeota and the closely related TACK sub-clade Thaumarchaeota discovered from their wide occurrence in ocean samples, which may have an even closer relationship with eucaryotes (Brochier-Armanet et al. 2008). The wider grouping of Crenarchaeota were originally thought to be extreme thermophiles, such as the aerobic Sulfolobus solfataricus found in an Italian hot pool growing at 80oC in a pH of 2-4. Notably this species has been found to have a form of epigenetic inhetirance implicating Rad54 dependent chromatin restructuring (doi:10.1073/pnas.1808221115). However since then ubiquitous low temperature Thaumarchaeote species, such as Crenarchaeum symbiosum and Nitrosopumilus maritimus have been discovered in cool oxic ocean. Nitrosopumilus is one of the smallest living organisms at 0.2 µm in diameter. It has a genome of 1.65 Mbp and lives by oxidizing ammonia to nitrite. Based on measurements of their signature lipids taken from ocean samples, These organisms are thought to be very abundant - estimated at 1028 cells in the world’s oceans (Konneke et al. 2005) - suggesting that they have a major role in global biogeochemical cycles and are one of the main contributors to the fixation of carbon. DNA sequences from Crenarchaea have also been found in soil and freshwater environments, suggesting that this phylum is ubiquitous to most environments. Significantly, these two organisms have been shown to possess a eucaryote type topoisomerase 1B (swivelase) which plays a major role in DNA replication and chromatin assembly in eucaryotes, and distinct from the 1B types found in some viruses and bacteria (Brochier-Armanet, Gribaldo & Forterre 2008). This confrims the founding eucaryote had a DNA genome. The closely related Caldiarchaeum subterraneum harbors a ubiquitin-like protein modifier system with structural motifs specific to eukaryotic system proteins. The presence of such a eukaryote-type system is unprecedented in prokaryotes, and indicates that a prototype of the eukaryotic protein modifier system is present in the Archaea (Nunoura et al. 2010). Thus the two cell types believed to be involved in eucaryote endosymbiosis are both closely related to existing ubiquitous species dominant on a global basis. Lokiarchaeota in particular which have been found in genetic analysis of a composite genome from around Loki's Castle, a black smoker vent in the Arctic, contain 175, (3.3%) genes that are very similar to eucaryote proteins, including actin. essential for phagocytosis (Guy & Ettema). This picture is supported by other living examples such as Candidatus Giganthauma karukerense cohabiting with γ-proteobacteria (fig 16b2).
 Fig 16a2b: Left: The inside-out hypothesis for the origin of the eucaryote cell has the original eocyte budding out from a cell comprising the nuclear envelope to form external blebs through the nuclear pore structures to facilitate symbiosis with γ-proteobacteria later forming the endoplasmic reticulum and engufing the mirochondria as the blebs evolved into a continuous external membrane. Centre: two archaeal Candidatus Giganthauma karukerense cells surrounded by ectosymbiotic γ-proteobacteria (Baum & Baum 2014). Right: Eucaryote fossil cell dated to around 1.45 billion years (Martin & Mentel 2010).
Fig 16a2b: Left: The inside-out hypothesis for the origin of the eucaryote cell has the original eocyte budding out from a cell comprising the nuclear envelope to form external blebs through the nuclear pore structures to facilitate symbiosis with γ-proteobacteria later forming the endoplasmic reticulum and engufing the mirochondria as the blebs evolved into a continuous external membrane. Centre: two archaeal Candidatus Giganthauma karukerense cells surrounded by ectosymbiotic γ-proteobacteria (Baum & Baum 2014). Right: Eucaryote fossil cell dated to around 1.45 billion years (Martin & Mentel 2010).
The second theory reverses the adaptive emphasis, placing it more firmly on the bacterium. There are several parasitic species of α-proteobacterium, which, after living internally in their host, kill their host to escape when it become vulnerable or compromized. Various bacteria such as Neisseria gonorrhoea utilise pore-forming porins to disrupt the host membrane, resulting in lysis, when the host cell becomes compromised. Eucaryote apoptosis, or controlled cell death, is mediated by the release of cytochrome-c and a network of related proteins amid a wave of oxidising free radicals through disruption of the electron transport chain, accompanied by porin-like pore proteins. All but one of the key components of this molecular network (excepting AIF, or apoptosis initiating factor) have a bacterial origin in the original mitochondrion, indicating the mechanisms of apoptosis originated with mitochondria, supporting the notion of an endoparasite that retained it's own control over the host cells ultimate fate (Lane 2005). Nick Lane has also suggested that this process may have played a role in driving the emergence of fusion sex as an adaption in which the non-freeliving mitochondrion now ensures survival of a compromised host by inducing host fusion and meiotic recombination. Cavalier-Smith (2002) has also suggested that cellular merging may have been common in the archaeal progenitor. Meiotic division would then emerge to compensate for polyploidy.
 Fig 16a3: Left: The hydrogen hypothesis proposes that LECA resulted from a versatilerespiring proteobacterium capable of metabolizing glucose to produce CO2 and H2 and an archaeal methanogen utilizing these to generate methane (Matin & Müller 1998). Right: Methanogens living inside the cytoplasm lining up beside hydrogenosomes in the marine ciliate Plagiopyla frontata illustrate the capacity of methanogens and hydrogenosomes to enter into a symbiotic metabolism (Lane 2005).
Fig 16a3: Left: The hydrogen hypothesis proposes that LECA resulted from a versatilerespiring proteobacterium capable of metabolizing glucose to produce CO2 and H2 and an archaeal methanogen utilizing these to generate methane (Matin & Müller 1998). Right: Methanogens living inside the cytoplasm lining up beside hydrogenosomes in the marine ciliate Plagiopyla frontata illustrate the capacity of methanogens and hydrogenosomes to enter into a symbiotic metabolism (Lane 2005).
The third approach (Lane 2005) places the emphasis on metabolic complementarity, an endosymbiotic relationship between an archaeote, such as a highly autonomous methanogen, able to generate all its metabolic components from basic molecules such as CO2 and H2, and a respiring bacterium containing hydrogenosomes which can generate precisely these same two molecules. This would set up a mutually beneficial energetic relationship which could lead quite naturally to the eucaryote emergence. Martin and Müller (1998) have proposed that mitochondrial symbiosis might emerge as an interaction under low oxygen between a respiring hydrogen-producing bacterium and an archaeal cell utilizing hydrogen to make ATP. Several anaerobic eucaryotes contain hydrogenosomes with double membranes and internal structure (Finlay & Fenchel 1989), which do precisely this. Some protista, such as Trichomonads and fungi such as Chritidiomycetae from cattle rumen have hydrogen-generating hydrogenosomes some of which share evolutionary homology with mitochondria (Martin & Mentel 2010). Some mitochondria in worms and molluscs are also able to shift to a low-energy anaerobic mode.
Many α-proteobacteria, including Rickettsiae (and related Wollbachia and Agrocbacterium), obligately live in the cytoplasm of other cells and so are naturally adapted to becoming an endo-symbiont of a glycolytic organism by providing respiring energy to the host's metabolism resulting in the mitochondrion. Giardia still retains traces of mitochondrial proteins so appears to have lost its respiring organelles, through specializing for an anaerobic parasitic habitat, rather than occupying a place in the tree before mitochondria were incorporated into eucarya (Adam 2000). In fact Giardia was found to have mitochondrial Fe-S proteins which showed up in vestigal mitochondria, now called a mitosomes (Zimmer 2009).
Having discovered the hydrogenosome, Martin and Müller (1998) suggested that the advent of mitochondrial symbiosis might be explained as an interaction under anoxic conditions between a respiring hydrogen-producing bacterium and an archaeal cell utilizing hydrogen to make ATP, as many archaea still do. Between 1000 and 600 Mya, the ocean underwent an anoxic period, possibly due to a snowball Earth phase which was broken by atmospheric CO2 buildup, and a major oxygen increase generated by cyanobacteria, which then precipitated the acidic ocean iron and triggered the Cambrian.
Some of the hydrogenosomes contain DNA, showing an evolutionary relationship with aerobic mitochondria. In particular, the hydrogenosomes of the anaerobic ciliate Nyctotherus ovalis, which thrives in the hindgut of cockroaches, have retained a rudimentary genome encoding components of a mitochondrial electron transport chain (Boxma et al. 2005). Similar correspondence of the hydrogenosome with mitochondria occurs in the proteome of Trichomonas vaginalis (Schneider et al. 2001). There is in fact a spectrum of mitochondria and mitochondrial remnants, called mitosomes, from oxygen producing aerobic through anaerobic mitochondria metabolizing nitrate or sulphate. Although there is debate about whether all these organelles share a single evolutionary origin, particularly mitosomes whose genes are entirely nuclear, hydrogenosomes and mitochondria share an overal evolutionary relationship. This sugests more versatile α-proteobacteria such as Rhodobacter, which are facultatively anaerobic photosynthetic bacteria could provide all these functions in a single mitochondrial progenitor. In an aerobic environment they produce CO2 and H2O, but in an anaerobic environment H2, CO2 and the organic acids succinate, propionate, formate and acetate (Sapp 2005). For discussion of the various research perspectives on this approach to eucaryote origins, see Martin & Müller (2007).
This massive increase in complexity remains obscure in the genetic and fossil records and requires some ingenious model construction to envisage how mitosis, meiosis, sexuality, the nuclear envelope, endoplasmic reticulum, cytoskeleton, and all the complexities of eucaryote regulation evolved. For a seminal work on this see (Cavalier-Smith 2010). Regardless of this, Lane and Martin's metabolic approach explains neatly why there is little sign of any of these structures in any existing prokaryote. In effect endo-symbiosis created a completely new energetic regime, in which the only niche players were the newly formed endo-symbiotic chimeras themselves, who then underwent a massive adaptive radiation to form ever more complex forms of cellular machinery and ultimately LECA and the diversity of eucaryotes as we now know them. There are echoes in this metabolic shangri-la of the conditions in lost city vents that we are coming to understand may have likewise given rise much earlier to LUCA.
 Fig 16a4; In 2015 a new Archaeal phyllum Lokiarchaeota, from the TACK super-phyllum, already known to include homologs of actins and tubulins and cell division sorting proteins (Guy & Ettema) illustrated in the tree, was discovered at the Arctic Mid-Ocean Spreading Ridge near the Loki's castle vent, having a closer evolutionary relationship with eucaryotes than any other known archeote, which also has the largest complement of ESPs, or eucaryote signature proteins, so far discovered (Spang et al.). The analysis of the genome of Lokiarchaeum revealed about 175 proteins that were related to eukaryotic proteins. These included actins, ubiquitin modifying proteins, diverse Ras-superfamily GTP-ases surpassed only by the eucaryote Naegleria gruberi (see below), an ESCRT gene cluster which eucaryotes is an essential component of the multivesicular endosome pathway for lysosomal degradation of damaged or superfluous proteins and plays a role in several budding processes including cytokinesis, autophagy and viral budding as well as of several additional proteins homologous to components of the eukaryotic multivesicular endosome pathway, including curvature sensing protein families involved in various aspects of vesicle/membrane trafficking or remodelling processes in eukaryotes.
Fig 16a4; In 2015 a new Archaeal phyllum Lokiarchaeota, from the TACK super-phyllum, already known to include homologs of actins and tubulins and cell division sorting proteins (Guy & Ettema) illustrated in the tree, was discovered at the Arctic Mid-Ocean Spreading Ridge near the Loki's castle vent, having a closer evolutionary relationship with eucaryotes than any other known archeote, which also has the largest complement of ESPs, or eucaryote signature proteins, so far discovered (Spang et al.). The analysis of the genome of Lokiarchaeum revealed about 175 proteins that were related to eukaryotic proteins. These included actins, ubiquitin modifying proteins, diverse Ras-superfamily GTP-ases surpassed only by the eucaryote Naegleria gruberi (see below), an ESCRT gene cluster which eucaryotes is an essential component of the multivesicular endosome pathway for lysosomal degradation of damaged or superfluous proteins and plays a role in several budding processes including cytokinesis, autophagy and viral budding as well as of several additional proteins homologous to components of the eukaryotic multivesicular endosome pathway, including curvature sensing protein families involved in various aspects of vesicle/membrane trafficking or remodelling processes in eukaryotes.
Just as we have investigated the enigmatic nature of LUCA, the last common ancestor of all life on earth, the nature of LECA, the last eucaryote common ancestor remains obscure. We have seen that certain archaea such as the Lokiarchaeota possess genes such as structural proteins which are signature of eucaryotes and may have enabled such species to engulf mitochondria. Eukaryotic signature proteins included several cytoskeletal components (actin homologues and gel-solin-domain proteins), ESCRT complex proteins and a wide variety of small GTPases that in eukaryotes are involved in various regulatory processes including cytoskeleton remodelling, signal transduction, nucleocytoplasmic transport and vesicular trafficking. We can now turn from the question of genetic fusion founding the eucaryotes to the question of what genetic and cellular regulation and signalling systems our common ancestor possessed and how early the key components enabling multi-cellular life evolved.
 Fig 16a4b: Ettema's team (Zaremba-Niedzwiedzka et al. 2017, Eme et al. 2023) have found a superphyllum comprising archaea in diverse environments including marine sediments, aquifers and hot springs which have a phylogenetic relationship with eucaryotes and include genes for vesicle formation, membrane-trafficking components and cyto-skeletal functions inuding ESCRT and TRAPP domains.
Fig 16a4b: Ettema's team (Zaremba-Niedzwiedzka et al. 2017, Eme et al. 2023) have found a superphyllum comprising archaea in diverse environments including marine sediments, aquifers and hot springs which have a phylogenetic relationship with eucaryotes and include genes for vesicle formation, membrane-trafficking components and cyto-skeletal functions inuding ESCRT and TRAPP domains.
The status of both the eocyte two domain model versus the three domain model and whether the Asgard phyllum represents a bridge from archaea to eucaryotes remains controversial (DaCunha et al. 2017, Watson T 2019).
Bacteria and eukaryotes have one set of lipids in their cell membranes, whereas archaeal membranes contain a different set (fig 1c1b). A mixture of the two was thought to be unstable. If eukaryotes came from archaea, they would have had to switch from using archaeal lipids to producing bacterial versions. The question of whether the archaeal and bacterial membranes are incompatible so that no hybridization could occur between their genomes has been resolved by the engineering (Caforio A et al. 2018) and discovery in the wild (Villanueva et al. 2018) of organisms containing both types of membrane component. The transitional organisms could thus have had such mixed membranes during the transition from archaea to eukaryotes.



Fig a4b2: Upper Left: Metabolic reconstruction of the Thorarchaeaota genomic bin (SMTZ1-83)(Seitz et al. 2016). Upper Right: Archaeal DNAof Methanothermus fervidus is spooled around histones like a slinky, which enables the selective opening up of individual genes for transcription, as opposed to the collective 'operons' of bacteria and enables the protection of DNA from extreme environments (Bowerman et al. 2021). Archaeal histones have evolutionary homology to eucaryote histones (Mattiroli et al. 2017). Lower: Syntrophy 'reverse flow model' (Spang et al. 2019) based on comparative analysis of the metabolic repertoire encoded by the various members of the Asgard archaea suggests that a metabolic syntrophy between anaerobic ancestral Asgard archaea and facultative anaerobic alpha-proteobacteria has provided the selective force for the establishment of a stable symbiotic interaction that has subsequently led to the origin of the eukaryotic cell. In this scenario, the archaeal progenitor generated reducing equivalents during growth on small organic substrates (for example, hydrocarbons and fatty acids) and the bacterial partner utilized these in the form of H2, small reduced inorganic or organic compounds, or by direct electron transfer. This contrasts with the hydrogen hypothesis of Martin and Muller, fig16a3, which suggests that a symbiosis between a strictly autotrophic hydrogen-dependent methanogenic archaeon and an H2-producing and CO2-producing alpha-proteobacterium led to the origin of the eukaryotic cell. It also contrasts with the previous syntrophic hypothesis proposed by Moreira and Lopez-Garcia, which invokes two bacterial and one archaeal partner(s) in the origin of the eukaryotic cell - first a syntrophic relationship established between a fermentative delta-proteobacterium and a hydrogen-dependent archaeal methanogen, which was incorporated into the cytoplasm of the bacterium through endosymbiosis. Subsequently, a second endosymbiosis event led to the uptake of a facultative aerobic alphaproteobacterium, which was suggested to have oxidized organic compounds and hydrocarbons produced by the host.
Although most members of these phylla are slow growing free-living organisms which are extremely difficult to culture in the lab, the first images of two such groups of organisms have been pictured by optical microscopy (Salcher et al. 2019).
t

Fig a4b3: Left: Images and evolutionary trees of key asgard organisms (Salcher et al. 2019). Right: Alternative Model of LECA emergence (Vesteg & Krajcovic 2011).
In 2019 Lokiarchaea, the first of the Asgard species to be discovered was finally successfully grown in culture. It had orginally been identified as a unique archaeal organism from microbial mud, dredged near Loki's Castle, a sea-floor hydrothermal vent field off the coast of Greenland. In a 2015 study in metagenomics, Ettema and his colleagues sequenced genetic fragments from the microbial portion in the sediment and assembled them into fuller genomes of individual species. One genome stood out. It was clearly a member of the archaea. But dotted throughout this genome were eukaryotic-like genes, named Lokiarchaea, after Loki, the trickster of Norse mythology (Lambert 2019).

Fig a4b4:(above) Images of Lokiarchaeota showing long branching membrane protrusions.
(below) {roposed changes in the symbiotic culture leading to the eucaryotes ((Imachi et al. 2019).
However, unbeknown to the metagenomics researchers, Hiroyuki Imachi and colleagues (Imachi et al. 2019) had been working since 2007 to cultivate microbes from deep-sea sediments. They built a bioreactor that mimicked the conditions of a deep-sea methane vent. Over 5 years, they waited for the slow-growing microbes in the reactor to multiply and then took samples placed these, along with nutrients, in glass tubes, which sat for another year before showing any signs of life. Genetic analysis revealed a barely perceptible population of Lokiarchaea. The researchers patiently coaxed the Lokiarchaea -- which took 2-3 weeks to undergo cell division -- into higher abundance and purified the samples. Over 12 years, in a breakthrough work, the researchers produced a stable lab culture (Prometheoarchaeum syntrophicum) containing only this new Lokiarchaeon and a different methane-producing archaeon in a symbiotic relationship. The researchers sequenced all the microbe's DNA, confirming that it does contain some genes that look like those found in eukaryotes. This has now enabled verification that the cultured genome contains the eucaryote-related genes from the metagenomics analyses and enables a much more retailed investigation of this critical group of prganisms.
Experiments with this single-cell organism suggest it usually - if not always - grows in association with another microbe that makes methane. Prometheoarchaeum breaks down amino acids for food and releases hydrogen, which feeds its partner. That methane-maker in turn helps Prometheoarchaeum thrive by chewing through the hydrogen, a buildup of which could otherwise cause even slower growth of Prometheoarchaeum. The complex partnership is another reason why the Asgard arcahaea are so hard to grow in the lab (Pennisi 2019).

Fig a4b4b: Candidatus Lokiarchaeum ossiferum (Rodrigues-Oliveira et al. 2022).
Insets: Evolutionary trees of ubitquitin genes UEV, E2, Vps22 and Vps25 (Hatano et al. 2022)
By carefully decanting cell cultures Rodrigues-Oliveira et al. (2022) have isolated a new Asgard Candidatus Lokiarchaeum ossiferum, which thrives anaerobically at 20°C on organic carbon sources. It divides every 7–14 days, reaches cell densities of up to 5 × 107 cells per ml and has a significantly larger genome compared with the single previously cultivated Asgard strain. Wu et al. (2022) isolated two other Asgard species—from rock collected from a hydrothermal vent in the Gulf of California to sequence their complete genomes. These harbored mobile pieces of DNA that contained bacterial genes involved in metabolism, suggesting these elements played a critical role in transferring genes among life's major branches. Hatano et al. (2022) have discovered four ubiquitin-ESCRT gene complexes eukaryotic cells use to bend, cut up, and stitch together their membranes to link internal compartments. On the other hand, Knopp et al. (2021) calculated that Asgard archaea contributed as little as 0.3% of the protein families believed to exist in the common ancestor of the eukaryotes, suggesting that the stresses on the host drove eucaryote complexification, such as the nucleus, Golgi apparatus, and the evolution of sex (Raval, Garg & Gould 2022).

Fig a4b5: Deep genomic research has uncovered hints of the earliest history of the FECA the eucaryote ancestor that preceded endosymbiosis between an archan cell and proetobacteria to form the mitochondria. A phase of incresead duplication of genes involved in cytoskeletal structures, translation, internal trafficking,replication, the cell cycle and protein modification occurred before the integration event that accompained endosymbiosis with a flood of genes involved in metabolism, implying that the archaean Asgard ancestor already possessed components of the internal membraneous architecture and information processing to support mitochondrial endosymbiosis.
By relatively timing events using phylogenetic distances, Vossberg et al. (2020) inferred that duplications in cytoskeletal and membrane-trafficking families were among the earliest events, whereas most other families expanded predominantly after mitochondrial endosymbiosis. Altogether, we infer that the host that engulfed the proto-mitochondrion had some eukaryote-like complexity, which drastically increased upon mitochondrial acquisition. This scenario bridges the signs of complexity observed in Asgard archaeal genomes to the proposed role of mitochondria in triggering eukaryogenesis.
Cyanobacteria enter the fossil record in the form of stromatolite mats around 3.5 billion years ago. William Schopf (Scientific American Feb 1991) found remnants of 3.6 billion-year-old stromatolites lying near fossils of 3.5 billion-year-old cells that resemble modern cyanobacteria, forming strings of putative microscopic cells. In 2016 (Nutman et al. 2016) discovered putative stromatolites dating to 3.7 billion years in the Isua formation, in Greenland. Thus oxygen-generating photosynthesis which provides an energetic basis for eucaryote respiratory metabolisms to survive arose very early.
Evidence for an increase in oxygen in the shallow oceans due to photosynthesis, based on analysis of the Manzimnyama Banded Iron Formation, South Africa goes back 3.2 billion years (Satkoski et al. 2015). Variations in selenium isotope concentrations in ancient rocks suggest that 2.32 billion to 2.1 billion years ago, shallow coastal waters held enough oxygen to support oxygen-hungry life-forms (Kipp et al. 2016). They note that recognition of oxygen-rich conditions in the early Paleoproterozoic opens up the possibility that there was a relatively long (∼200 My) interval that may have been favorable for the evolution of complex life forms long before the fossil record indicates their rise to ecological importance. This timing corresponds to the so-called great oxygenation event, crisis or catastrophe, in which molecular O2 entered the atmosphere for the first time, after a long period where any oxygen released was taken up by oceanic iron and other terrestrial minerals entering the oxidized state, resulting in an approximate doubling of mineral diversity. The centimeter-scale structures in the ∼2.1 Ga Francevillian Series of Gabon would be consistent with this interpretation. But other fossil and evolutionary evidence for eucaryote cells doesn't appear convincingly until around 1.6 billion years ago, when fossils resembling crown red algae have been found, indicating eucarytoes welll already well diversified into multicellular forms (Bengtson 2017). By comparison, eukaryotic microfossils containing mitochondria go back only 1.45 Bya in the fossil record (Martin & Mentel 2010 - fig 16b2).
Thus an intriguing idea about the trigger for eucaryote emergence is that the rise in oxygen levels, in the great oxygenation event drove the transformation of an archaeal lineage into forming the first eukaryotes, through a combination of much greater respiratory yields arising  from mitochondrial endosymbiosis in an oxygenated atmosphere and increased genetic stresses due to reactive oxygen species. Selective pressure for efficient repair of ROS/UV-damaged DNA then drove the evolution of sex, which required cell-cell fusions, cytoskeleton-mediated chromosome movement, and emergence of the nuclear envelope. The model implies that evolution of sex and eukaryogenesis were inseparable processes (Gross & Bhattacharya 2010). This idea places a central focus on processes of cell fusion, DNA repair and recombination in archaea. Although such processes have only recently begun to be explored in archaea, there are several distinctive examples of cell fusion and conjugation, along with recombination in response to genetic stress among the archaea (Sowers & Schreier 1999, Wagner et al. 2017).
from mitochondrial endosymbiosis in an oxygenated atmosphere and increased genetic stresses due to reactive oxygen species. Selective pressure for efficient repair of ROS/UV-damaged DNA then drove the evolution of sex, which required cell-cell fusions, cytoskeleton-mediated chromosome movement, and emergence of the nuclear envelope. The model implies that evolution of sex and eukaryogenesis were inseparable processes (Gross & Bhattacharya 2010). This idea places a central focus on processes of cell fusion, DNA repair and recombination in archaea. Although such processes have only recently begun to be explored in archaea, there are several distinctive examples of cell fusion and conjugation, along with recombination in response to genetic stress among the archaea (Sowers & Schreier 1999, Wagner et al. 2017).
Fig 16a4c: Model of evolution of archaea and bacteria from a central complexifying eucaryote pathway.
An alternative theoretical approach advanced by Forterre (2013) is that LUCA rather than LECA forms the founding archetype of eucaryotes, with both archaea and bacteria diverging from a central complexifying pathway by reductive evolution in which specialization e.g. for high temperatures, or specific metabolic pathways resutls in a strategic simplification of evolving genomes. However this lacks an explanation for the energetic principles involved in the endosymbiosis as a founding trigger fo complexity, or how a complex possibly nucleated, organism could have evolved in the absence of clear prevailing energetic limitations.
There has also been a considerable degree of horizontal gene transfer between bacteria and archaea, that has contributed to the ecological diversity of modern archaeal species, with five times as much DNA being transferred into archaea than the reverse, providing metabolic genes for new niches. Haloarchaea, which are oxygen-respiring heterotrophs, originated from methanogens, which are strictly anaerobic hydrogen-dependent autotrophs. A recent study suggested that a single HGT event resulted in the transfer of a large piece of DNA that contained approximately 1,000 bacterial genes to a methanogen, which became the last haloarchaeal common ancestor (Wagner et al. 2017).

Fig 16a4d Left: Aggregation of Sulfolobus cells concomitant with Ced-based DNA exchange under UV stress, Centre: Bi-directional conjugation in Haloarchaea.
Right: Conjugation in Solfolobus.
As is the case with bacteria and eucaryotes, viruses can be integrated into archaeal genomes, however very few have been shown to have a cost to their hosts. Sulfolobus spindle-shaped virus 1 has been shown to produce virus particles that bud from host cells without killing them in a manner that is very similar to the budding process of eukaryotic enveloped viruses. Several thermophilic archaea have been found to release membrane vesicles through budding. Membrane vesicles appear to have a protective function and enable intercellular DNA transfer and recombination without a viral infection. In crenarchaeal species, endosomal sorting complex required for transport III (ESCRT-III) homologues mediate the release of membrane vesicles. ESCRT-III proteins facilitate membrane scission in eukaryotes and members of the TACK phylum (ibid).
Following DNA damage, Sulfolobus cells aggregate and chromosomal DNA is transferred between the cells involving type IV pilii, which presumably provides an intact DNA template for homologous recombination. A group of proteins (Ced) have also been isolated that are not related to the pili, but are essential for DNA transfer (van Wolferen et al. 2016).Here the pili appear to anchor the cells to one another and the Ced system transfers DNA directly through the adjacent cell membranes. DNA is exported from the donor cell after cell-to-cell contact is initiated. DNA is then actively imported by the Ced system. Incoming DNA can be used as a template for homologous recombination to repair damaged DNA. A contact-dependent DNA import system has thus far not been observed in other archaea, or in bacteria or eukaryotes. Several conjugative plasmids have also been isolated exclusively in members of the family Sulfolobaceae, suggesting that conjugative plasmids might be limited to this family (Prangishvili et al. 1998).
DNA transfer between Halobacterium volcanii cells was shown to be bi-directional and dependent on cell-cell contact (Rosenshine, Tchelet & Mevarech 1989). Many haloarchaeal species form biofilms, and frequent DNA exchange occurs in H. volcanii bio-films. Cytoplasmic bridges maintain the cytoplasmic continuity of two connected cells enabling the transfer of DNA between cells. As multiple cytoplasmic bridges were shown to connect two cells at the same time, classical bacterial conjugation was ruled out as a transfer mechanism. This was supported by the finding that both chromosomal and plasmid DNA were exchanged at similar frequencies. Hetero-diploid cells are formed temporarily during this process. These cells contain the chromosomal and plasmid DNA of both parental strains. Recombination between the chromosomes and subsequent segregation will occasionally lead to a hybrid of the parental strains.

Fig 16a5: (Left) Complement of signalling systems found in Naegleria gruberi (Fritz-Laylin et al. 2010), a free-living single celled bikont amoebo-flagellate, belonging to the excavata, which include some of the most primitive eucaryotes such as Giardia and Trichomonads. Nevertheless it is capable of both oxidative respiration and anaerobic metabolism and can switch between amoeboid and flagellated modes of behavior, regenerating complete centrioles and flagellae de novo (Fritz-Laylin & Cande 2010). The Naegleria genome sequence contains actin and microtubule cytoskeletons, mitotic and meiotic machinery, suggesting cryptic sex, several transcription factors and a rich repertoire of signalling molecules, including G-protein coupled receptors, histidine kinases and second messengers including cAMP. One strain analyzed is a composite of two distinct haplotypes indicating hybridization. Although sexual mating has not been observed in Naegleria, the heterozygosity found in its genome is typical of a sexual organism, with perhaps infrequent matings. Additionally, identification of the core RNAi machinery indicates that Naegleria may use this mechanism. Nagleria fowlerii is a notorious warm-water species causing meningitis. (Right) Three of four genes involved in meiosis, HOP1 believed to be a component of the synaptonemal complex essential for sexual recombination, MRE11 meiotic recombination 11 involved in recombination, telomere processing and double-stranded DNA break repair and RAD51 also involved in meiosis and double stranded DNA repair. All three show a wide evolutionary distribution across eucaryotes including Giardia, implying that LECA the last eucaryote common ancestor was a sexual organism (Ramesh, Malik & Lodgson 2005). MRE11, along with RAD51 also show homology with Archaea, suggesting an even deeper origin of sexuality.
The fact that primitive eucaryotes such as Giardia and Trichomonads appear to lack key structures and processes such as mitochondria and sexual recombination initially led to the notion that the founding eucaryote was a simple amoeboid cell lacking the genetic complexity of more advanced protista and higher organisms. However it is now clear that a tree reconstruction artefact, known as Long Branch Attraction, is responsible for the apparent early emergence of the fast evolving Archezoa in the eukaryotic tree. The notion that all extant eukaryotes are ancestrally mitochondrial is strongly supported by the discovery of rudimentary mitochondrial organelles in all analysed Archezoa. Part of the problem comes from the fact that organisms such as Giardia and Trichomonads have specialized for parasitic microenvironments and in particular anaerobic conditions where they can suffer rapid loss of inessential genes, distorting the evolutionary picture. When we look at close free-living relatives of these organisms, we discover a complex genetic complement containing most of the functionalites of "advanced" eucaryotes.
Naegleria gruberi transitions from amoeboid to flagellate behavior and back (https://youtu.be/yy5zI3LBK24).
The free-living amoebo-flagellate Naegleria gruberi for example (Fritz-Laylin et al. 2010) belongs to a varied and ubiquitous protist clade (Heterolobosea) that diverged from other eukaryotic lineages over a billion years ago and is very close to the hypothetical root of the eucaryote tree (see fig 16b5). The Naegleria genome, analyzed in the context of other protists, reveals a remarkably complex ancestral eukaryote with a rich repertoire of cytoskeletal, sexual, signaling, and metabolic modules.
 Fig 16b: Consensus tree for eucaryote root (Brinkmann & Philippe 2007). The assumption underlying Woese's paradigm - that simple organisms (e.g., prokaryotes and Archezoa) represent genuinely primitive intermediates in the progressive construction of complex eukaryotic cells - has on the basis of more recent genetic analysis been replaced by that of a complex last common eucaryote ancestor (LECA/LCAEE). In the consensus picture, any feature present in some opisthokonts (e.g., animals) and in some bikonts (e.g., plants) is necessarily ancestral, i.e., inherited from the LECA. This implies that the LECA was in Brinkman and Phillipe's words: "amazingly more complex than previously thought. Among others, the LCAEE already possessed mitochondria, an efficient cytoskeleton associated with several intracellular transport systems, an endo-membrane system interconnected by a complicated vesicular transport machinery, including an endoplamic reticulum, a Golgi apparatus, a standard nucleus, efficient secretory and uptake pathways, recycling of food‐vacuoles, peroxisomes, spliceosomal introns, and flagella‐dependent motility. Therefore, simple extant eukaryotes have evolved from a complex LCAEE mainly by loss and secondary simplification".
Fig 16b: Consensus tree for eucaryote root (Brinkmann & Philippe 2007). The assumption underlying Woese's paradigm - that simple organisms (e.g., prokaryotes and Archezoa) represent genuinely primitive intermediates in the progressive construction of complex eukaryotic cells - has on the basis of more recent genetic analysis been replaced by that of a complex last common eucaryote ancestor (LECA/LCAEE). In the consensus picture, any feature present in some opisthokonts (e.g., animals) and in some bikonts (e.g., plants) is necessarily ancestral, i.e., inherited from the LECA. This implies that the LECA was in Brinkman and Phillipe's words: "amazingly more complex than previously thought. Among others, the LCAEE already possessed mitochondria, an efficient cytoskeleton associated with several intracellular transport systems, an endo-membrane system interconnected by a complicated vesicular transport machinery, including an endoplamic reticulum, a Golgi apparatus, a standard nucleus, efficient secretory and uptake pathways, recycling of food‐vacuoles, peroxisomes, spliceosomal introns, and flagella‐dependent motility. Therefore, simple extant eukaryotes have evolved from a complex LCAEE mainly by loss and secondary simplification".
Recently, a molecular dating study based on a large phylogenomic dataset with a relaxed molecular clock and multiple time intervals yielded in a surprisingly recent time estimate of 1085 Mya for the origin of the extant eukaryotic diversity. Therefore, extant eukaryotes seem to be the product of a massive radiation that happened rather late, at least in terms of prokaryotic diversity (Brinkmann & Philippe 2007). Given the large number of novel eucaryote genes and folds(Lane & Martin 2010) this late diversification needs to be carefully rationized with the results of (David and Alm 2010) shown in fig 10 and with the fact that the cyanobacterial chloroplast was incorporated by plants between 1200 and 1800 Mya (Dorrel & Howe 2012).
The Eucaryote Dawn
The conflict between early eucryote fossil evidence dating back only 800 mya and molecular evolution dating back nearly to 2 bya has now been resolved through the discovery of eucaryote protosteroids in rocks from Australia spannnig the early phase (Brocks et al. 2023).

Fig 23b: Geological time chart comparing the molecular fossil, microfossil and phylogenetic records of early eukaryote evolution. a, Relative abundances of aromatic protosteroids (purple and cyan tones) and crown- group steroids (reds, blues and greens), highlighting the transition from a protosterol biota to a 'crown-sterol biota' in the Neoproterozoic era. Each horizontal colour bar represents one sample, and grey triangles assign data bundles to geological units 1 to 11 (key provided in Methods). Details on data assembly and geological formations (1–11) are provided in Methods. Ga, billion years ago. b, Phylogenetic tree of the domain Eukarya with black, red and green highlighting crown-group branches. Stem-group branches (purple) are hypothetical only, illustrating the notion that mid-Proterozoic ecosystems may have been dominated by extinct stem forms that did not produce crown sterols. LECA, the last common ancestor of all extant eukaryotes, may have emerged between 1.2 and more than 1.8 Ga. c, Microfossils of early eukaryotes: 1–5, likely crown-group Eukarya (approximately 1.1 to 0.7 Ga); 6–11, microfossils that are possibly or certainly eukaryotic but lack diagnostic crown-group characteristics (1,600 to 1,000 Ma). Detailed information and image credits are provided in Methods. C, Cenozoic era; Cr, Cryogenian period; Ed, Ediacaran period; M, Mesozoic era; P, Palaeozoic era; Palaeo., Palaeproterozoic era; Tn, Tonian period. For images in c: image 1, Melicerion poikilon, a possible testate rhizarian54, image courtesy of S. Porter, reproduced with permission from ref. 54; image 2, Bonniea dacruchares, a testate amoebozoan54, image courtesy ofmS. Porter, reproduced with permission from ref. 54; image 3, Bangiomorpha pubescens, a likely bangiacean rhodophyte alga55,56, image courtesy of N. Butterfield, reproduced with permission from ref. 57, © The Palaeontological Association; image 4, Proterocladus antiquus, a likely multicellular, benthic, siphonocladalean chlorophyte alga11, image courtesy of Q. Tang; image 5, Ourasphaira giraldae, a likely fungus12, image courtesy of C. Loron; image 6, Trachyhystrichosphaera aimika, a microfossil with diagnostic eukaryotic features, image courtesy of J. Beghin, reproduced from ref. 58 with permission from Elsevier; image 7, Leiosphaeridia jacutica, a microfossil of possible eukaryotic origin, image courtesy of J. Beghin, reproduced from ref. 58 with permission from Elsevier; image 8, Satka favosa, a microfossil with diagnostic eukaryotic features9, image courtesy of E. Javaux, reproduced from ref. 9; image 9, Valeria lophostriata, a microfossil with diagnostic eukaryotic features9, image courtesy of E. Javaux, reproduced from ref. 9; image 10, Tappania plana, a microfossil with diagnostic eukaryotic features1, image courtesy of Y. Leiming, reproduced with permission from ref. 1, © The Palaeontological Association; image 11, Shuiyousphaeridium macroreticulatum, a microfossil with diagnostic eukaryotic features1, image courtesy of Y. Leiming, reproduced with permisison from ref. 1, © The Palaeontological Association.
The eucaryote tree of life remains an enigma, particularly in terms of locating the root of the tree. Originally portrayed in terms of the five kingdoms of plants, animals and fungi, protruding from the single-celled protista, complementing the eubacteria and archaeotes classed as monera. However the tree has now been revised (Adl 2012). It is now recognized that multi-cellularity has arisen independently in many branches of the eucaryotes (Brown et al. 2012).

Fig16b3: (Left) Traditional five-kingdom eucaryote tree of life (Whittaker). (Right) Multiple evolution of multcellularity in diverse eucaryote super-groups
extends to many groups besides fungal and animal Opisthokonts and plant and algal Archaeplastida (Brown et al. 2012).

Fig16b3b: A new entire supergroup of carnivorous eucaryotes discovered in 2022 (Tikhonenkov et al. 2022).
However a key step in multicellularity has been linked to a single critical mutation. To form and maintain organized tissues, cells must coordinate how they divide relative to the position of their neighbours. One important aspect of this process is  orientation of the mitotic spindle, a structure inside the dividing cell that distributes the chromosomes —and the genetic material they carry — between the daughter cells. When the spindle is not oriented properly, malformed tissues and cancer can result. In a diverse range of animals, the orientation of the spindle is controlled by an ancient scaffolding protein that links the spindle to “marker” proteins on the edge of the cell. Anderson et al. have now used a technique called ancestral protein reconstruction to investigate how this molecular complex evolved its ability to position the spindle. First, the amino acid sequences of the scaffolding protein’s ancient progenitors, which existed before the origin of the most primitive animals on Earth, were determined.Anderson et al. (2015) computationally retraced the evolution of large numbers of present-day scaffolding protein sequences down the tree of life, into the deep past. Living cells were then made to produce the ancient proteins, allowing their properties to be experimentally examined. By dissecting successive ancestral versions of the scaffolding protein, they deduced how the molecular complex that it anchors came to control spindle orientation. This new ability evolved by a number of 'molecular exploitation' events, which repurposed parts of the protein for new roles. The progenitor of the scaffolding protein was actually an enzyme, but the evolution of its spindle-orienting ability can be recapitulated by introducing a single amino acid change that happened many hundreds of millions of years ago.
orientation of the mitotic spindle, a structure inside the dividing cell that distributes the chromosomes —and the genetic material they carry — between the daughter cells. When the spindle is not oriented properly, malformed tissues and cancer can result. In a diverse range of animals, the orientation of the spindle is controlled by an ancient scaffolding protein that links the spindle to “marker” proteins on the edge of the cell. Anderson et al. have now used a technique called ancestral protein reconstruction to investigate how this molecular complex evolved its ability to position the spindle. First, the amino acid sequences of the scaffolding protein’s ancient progenitors, which existed before the origin of the most primitive animals on Earth, were determined.Anderson et al. (2015) computationally retraced the evolution of large numbers of present-day scaffolding protein sequences down the tree of life, into the deep past. Living cells were then made to produce the ancient proteins, allowing their properties to be experimentally examined. By dissecting successive ancestral versions of the scaffolding protein, they deduced how the molecular complex that it anchors came to control spindle orientation. This new ability evolved by a number of 'molecular exploitation' events, which repurposed parts of the protein for new roles. The progenitor of the scaffolding protein was actually an enzyme, but the evolution of its spindle-orienting ability can be recapitulated by introducing a single amino acid change that happened many hundreds of millions of years ago.
Fig 16b3b: Tree of Dlg protein evolution to animal multicellular mitotic division (Anderson et al. 2015)
Closer studies of the genetic and phylogeny of a large number of eucaryote proteins has led to a revision of the tree in which single-celled species such as amoebae are grouped with animals and fungi in the opisthokonts, which are separated to the ery root of the tree from multi-cellular plants forming the archaeplastids. Animals are closer cousins to single-celled choanoflagellates than to other multi-cellular organisms, such as fungi and plants. Giant kelp are closer relatives to single-celled diatoms than to multicelled red seaweeds or plants. Likewise separate groups consisting of Rhizaria, Alveolates and Stametopiles have been found to form a single supergroup now called SAR.


Fig 16b4: Left: Eucaryote evolutionary tree showing the Hemimastigophora as a novel supra-kingdom-level lineage (Lax et al. 2018) Right: Consistent rooted eucaryote tree using two phylogenetic measures based of eubacterial-origin eucaryote gene sets. Malawimonas jakobiformis and Naegleria gruberi are illustrated to show common features close to the root of the tree.
Attempts to find the root of the eucaryote tree naturally revolve around linking eucaryotes to their founding archaeota using RNAs and proteins linked to nucleic acid processing and other processes inherited from the founding archaeal genome. However, the archaeal sequences differ substantially from their eukaryotic counterpart, resulting in extremely long phylogenetic distances between archaea and eukaryotes. The use of a distant outgroup in phylogenetic reconstruction is highly problematic because the remaining phylogenetic signal is very weak, and so correct positioning of the root is even weaker, creating a nonphylogenetic signal that is often stronger than the phylogenetic signal, thereby favoring long-branch attraction so that fast evolving eukaryotes are constantly found at the base of all other eukaryotes.


Fig 16b5: Left: Revised eucaryote tree (Milius 2015) taking into account supergroupings which include both single-celled and multicelled organisms rooted as closely as, possible given several lines of phylogenomic evidence (Burki 2014) . A second phylogenetic tree has additional branches. Right: A newer revised supergrouping (Burki et al. 2020).
Rare cytological and genomic changes specific to some eukaryotic lineages have also been considered for rooting of the eukaryotic tree, but an innovative alternative strategy is to use the eubacterial genes which have been incorporated into eucaryote lines, particulaly the alpha-proteobacterial genes originally incorporated with the mitochondria. By combining the "ALPHA-PROT" dataset using of 42 eukaryotic proteins with a mitochondrial function encoded by the nuclear or mitochondrial genomes as phylogenetic markers with the "EUBAC" dataset using 37 eukaryotic genes acquired by ancient lateral gene transfers from different eubacterial sources and including a wider set of newly sequenced species the analysis arrives at the following consistent rooted tree (Derelle et al. 2015). This places malawimonads such as Malawimonas jakobiformis and discoba such as Naegleria gruberi on opposite sides of the root (fig16c), despite the act that they are both classed as excavates (Cavalier-Smith 2010, 2014) which are otherwise regarded as monophyletic based on their flagellar ultrastructure.


Fig 16c: Leftt: The early origin of sexuality is attested to by recent research into the extended family tree of amoebae, which shows that sexualtiy is likely to have arisen in the common ancestor and been subsequently lost in asexual protist species, rather than the reverse (Lahr et al 2011). Amoeboid organisms (left) highlighted on the eucaryote tree of life. SAR indicates the group composed of Stramenopila, Alveolata and Rhizaria. Two principal branches (right) unikont amoebozoa and dikont rhizaria show confirmed (black), direct evidence such as meiosis (grey) and indirect evidence (white) for sexualty showing it is a likely founding characteristic. Evidence for sex in Giardia (Poxeleitner 2008, Birky 2004, 2009, Xu et al. 2012) supports the idea that sexuality arose in the last common ancestor of all eucaryotes, very early in evolution (Lane 2009a). Right: Distribution of (meiotic) sex and selected sex-related features in eukaryotes (Speijer et al. 2015). Black boxes, well-documented sex; gray boxes, limited evidence for sex (rare direct observations, indirect inference from genomic data); no boxes, no published evidence. Absence of boxes does not directly imply absence in a lineage, especially for those given in gray, with limited (or absent) genome-scale sequence data. HAP2 and GEX1 are two proteins functioning in cell and nuclear fusion, respectively. A-F: Representatives of deep eukaryotic lineages without published evidence for sex thus far. (A) Picomonas judraskeda (Picozoa). (B) Andalucia incarcerata (Jakobida). (C) Ancyromonas sigmoides (Ancyromonadida). (D) Roombia truncata (Katablepharida). (E) Breviata anathema (Breviatea). (F) Telonema subtilis (Telonemia). (G) An undescribed malawimonad. However, Jakobida, Glaucophyta, and Malawimonadida, thus far unreported to exhibit sex, all contain genes involved in plasmogamy (gamete fusion) and/or karyogamy (nuclear fusion).
Contrary to the idea that sexuality and sexual recombination are late adaptions of advanced eucaryotes, investigation of meiosis-related genes HOP1, MRE11, MLH/PMS and RAD51/DMC1, as shown in fig 16a5, produces evolutionary trees showing the occurrence of these genes across the eucaryotes from Giardia to Homo, implying sexuality is a founding characteristic of all extant eucaryotes (Ramesh, Malik & Lodgson 2005). Two of these trace a genetic ancestry back to archaea, supporting an even deeper origin for recombination. Lahr et al (2011) state on the basis of diverse evidence including genetic evidence of meiosis, that amoeboid forms of both unikont amoebozoa and dikont rhizaria show trends consistent with founding sexuality, later lost in some branches. In their words, mapping sexuality onto the eukaryotic tree of life demonstrates that the majority of amoeboid lineages are, contrary to popular belief, anciently sexual, and that most asexual groups have probably arisen recently and independently. Speijer et al. (2015) state that eukaryotic sex is extremely wide-spread and already present in the last eukaryotic common ancestor and the general mode of existence of eukaryotes is best described by clonally propagating cell lines with episodic sex triggered by external or internal clues due to genetic or environmental stress. Pertinent here is that multicellular organisms are precisely a chain of cells propagated by mitosis, albeit physically joined, with intermittent sexual fertilization occurring in the alternation of the generations. Based on these considerations, they consider that eukaryotic sex likely developed as a cellular survival strategy, in the context of internal reactive oxygen species stress generated by the (proto-) mitochondrion, also inducing structures such as the nuclear envelope.
Fig 16c2 below shows the prevalence of sperm-ovum fusion throughout the animals, core plant species and other single and multi-celled eucayrote branches. Lane (2005) proposes that early in the evolution of endosymbiosis, the mitochondrion evolved mechanisms of cellular merging and meiosis to avoid becoming trapped in a defunct archaeal cell. Sexuality does appear to be universally driven by mitochondrial competition, resulting in a uni-parental inheritance of cytoplasmic organelles. This in turn results in a symmetry-breaking between a large ovum and a sperm contributing, with some accidental exceptions, only nuclear DNA to fertilization. Mitochondria, like endosymbiotic Wollbachia in insects, can also induce male cytoplasmic sterility to hijack the sex ratio in their favour. There is some heteroplasmy (paternal mitochondrial leakage) in 10-20% of humans, leading to diverse mitochondrial diseases and 20% of 295 angiosperm species have been found to have some degree of bi-parental inheritance. Thus 7.5% of European angiosperms are gynodioecious - consisting of both hermaphrodites and females.
Fig 16c2: Life cycles of representative single celled and multicellular eucaryotes show that all branches have clear evidence of sexual reproduction. (Lane 2005) proposes that sexuality is driven by mitochondrial competition resulting in a uni-parental inheritance of cytoplasmic organelles. This in turn results in oogamy, a symmetry-breaking between a large ovum and a sperm, transmitting with some accidental exceptions, only nuclear DNA to fertilization. This pattern of sperm-ovum fertilization is widely spread on the eucaryote tree.
This pattern of sperm-ovum fertilization is widely spread on the eucaryote tree (fig 16c2), spanning choanoflagellates (Levin & King 2013, Wosnica et al. 2017) through animals and founding fungi, all plants, from the founding charophyta (see also fig 26a) and their sister chlorophytes such as Volvox, through Bryophyta (mosses and liverworts) to ferns, cycads and ginkos and is likewise evident in both the Rhizaria and Alveolata. Animals have a variety of lifes cycles, sometimes involving parallel cycles of parthenogenesis and sexuality (Daphnia), larval and adult forms (Cnidaria), as well as the standard sexual cycles shared by mammals (Human) and choanoflagellates. Fungi utilize a variety of sexual cycles. Chytrids, parasitic fungi notorious for wiping out amphibians, lie right at the root of the fungal tree and have male and female motile gametes confirming their relationship with metazoa. Higher fungi having a pecking order of compatability types and exchange nuclei by conjugation, rather than fusion sex. Schizophyllum commune for example has 28000 mating types via incompatibility genes on 2 chromosomes, each with some 300 and 90 variants respectively (Lane 2005). Both the higher fungi and red algae have complicated three stage life cycles involving sexual fertilization and subsequent mitotic phases, with haploid, di-karyotic and diploid stages. Higher plants retain the symmetry-broken form of sperm-ovum fertilization adapted to land living, via pollen as male gametes internally fertilizing an ovun to form a seed. Organisms that produce isogametesand practice isogamy, such as the sea lettuce Vulva and the single celled alga Chlamydomonas have to endure massive cytoplasmic genetic warfare. In the case of Chlamydomonas rheinhardtii, 95% of the mitochondrial DNA is destroyed by the opposing organelle DNA in the process. In the polynuclear slime mould Phaesarium polycephalum, which has 13 merging sexes with a strict pecking order and fertilizes by isogamete fusion, a mitochondrial plasimid mF facilitates fusion and recombination in mitochondria (Sakurai et al. 2004). The fungus Neurospora crassa, by contrast appears to destroy the paternal (transmitting) mitochondria over several days in favour of the maternal (accepting) mating type (Lee &, Taylor (1995). In yeasts which also have fusion sex, recombination occurs between the mitochondrial genomes inherited from both parents (Frisch et al. 2014). Mitochondrial recombination in these species enables optimal selection for respiratory efficiency and low mutation levels. Among the Stramenopiles, the brown alga Laminaria has sperm-ovum oogamy and oomycota and diatoms display isogamy. Among the Amoebozoa in addition to Physarum, Myxogastria and Dictyosphaeria also exhibit isogamy. Microsporidia contain a mix of sexual and asexual species, but genetic analysis implies repeated loss of sexuality from a sexual founding species (Ironside 2007, Lee et al. 2009). Many single-celled species exhibit a combination of asexual and sexual reproduction, in which sexual reproduction is intermittent, e.g. under environmental stress or DNA damage, and is thus hard to verify. Such species can also lead to asexual clones due to a recessive mutation losing intermittent sexuality. Many species are difficult or impossible to culture and indirect evidence may be inferred from genetic analysis indicating meiotic recombination of allies. Radiolaria have evidence of a synaptonemal complex (Lecher 2011), meiosis and a zygote (comenius.susqu.edu/biol/202/chromalveolata/rhizariae/radiolaria/). Even the excavata (fig 16c2 lower right) have evidence of cryptic sexual recombination, likely to occur intermittently under stress amid asexual reproduction, with Giardia showing evidence of recombination between its two resident genomes (Poxleitner et al. 2008, Birky 2009, Xu et al. 2012), Trypanosomes showing immuno-flourescent evidence of recombination of red and green emitting strains in vivo to form yellow recombinants (Hide 2008), and Naegleria and Tetramitus species also showing evidence of intermittent sexuality (Pernin et al. 1992, Fritz-Laylin et al. 2011).
Plants and animals each made the leap to multicellularity just once, but fungi likely evolved complex multicellularity in the form of fruiting bodies - think mushrooms - on about a dozen separate occasions, based on a review of how different species of fungi - some single-celled, some multicellular - are related to one another. Likewise red, brown, and green algae all evolved their own multicellular forms over the past billion years or so. Genetic comparisons between simple multicellular organisms and their single-celled relatives have revealed that much of the molecular equipment needed for cells to band together and coordinate their activities may have been in place well before multicellularity evolved. The protist Capsaspora uses some of the same molecules as animals to turn genes on and off at particular times and places: protein transcription factors and long strands of RNA that don't encode proteins, but its promoters - the regulatory DNA that interacts with transcription factors - were much shorter and simpler than in animals, illustrating how existing genes can be harnessed to make more complex organisms (Pennisi 2018).


Fig 16c3: Left: Origin of multicelled animals arises from single-celled Opiskonts developing genetic mechanisms for sequential cell states as part of social adaption. These sequential cell states then become the sequential mechanisms of stem cells in multicelled animals. By contrast with the notion of the first multicelled animals being simple balls of choanoflagellate-like cells, the transcriptome information suggests the sponge archaeocyte transcriptome (lower left) provides a closer link taking the role of a primordial stem cell that then becomes key to the structured development of multicellular body plans, including that of the sponge involving three interacting cell types (Sogabe et al. 2019). Right: Choanoeca flexa, a new species of choanoflagellate, crowds together to form groups of many individuals. The organisms form a ball shape with their tail-like flagella pointing out to help with swimming, but can quickly switch to a relaxed sheet shape with their flagella pointing in when feeding. The GIF shows C. flexa transitioning from swimming mode to feeding mode, triggered by light via rhodopsin (doi:10.1126/science.aay2346)..
A detailed evolutionary picture showing the relationship between metazoa and protozoans and the bridge between choanoflagellates and sponges has been developed in association with elucidating the genome of the single-celled choanoflagellate Monosiga brevicollis (King et al. 2008).
A team led by Sandie and Bernard Degnan has since shown the transition to multicellularity appears to depend on stem-cell like sequential changes in single-celled Opiskonts lying more deeply in the evolutionary tree than Chonanoflagellates, as noted in fig 16c3 (Sogabe et al. 2019). Essentially the picture is that temporal changes in gene expression among single celled species resulting in differing cell types during their life cycle, for example transitioning between free-swimming and gathering in colonial forms, led to multicellularity, when components of the temporal sequence became spatial expression of coexisting differentiated cell types, with stem cells forming the progenitors of differentiation.

Fig 16c4: Transition from choanoflagellate to sponge (Naumann & Burkhardt 2019)
Naumann & Burkhardt (2019) have since found that the cells in a choanoflagellate colony are not all identical and differ in morphology and the ratio of their organelles, suggesting that spatial cell differentiation was already happening in the choanoflagellate lineage, and perhaps even earlier - a possibility that blends the ideas of the Degnan team, that the capacity for differentiation is ancient and the transition to animal multicellularity was gradual, with the evolutionary sequence that this could have resulted in the metazoa via choanoflagellate-like cells.
 Fig 16d: (a) Tree of organisms expressing various forms of integrin-related proteins, from α-actinin to α-integrin includes diverse branches of single-celled eucaryotes (Sebe-Pedros et al. 2010). α-actinin was expressed in all branches. Both α- and β-integrin are expressed in those marked with (*) including single-celled Capsaspora owcarzaki and Amastigomonas sp. (b) Tree of tyrosine kinases (Suga et al. 2012). Cytoplasmic CTKs have a deep eucaryotic origin while receptor RTKs evolve later. (c) Tree of GPCR components extends back to LECA (Mendoza et al. 2014) making this signalling pathway, central to the nervous systems of higher animals, a founding unit of eucaryote evolution. All except those marked with * had one or more GPCR components and all clades included species possessing canonical GPCR, across both unikonta and bikonta, implying a common origin with LECA.
Fig 16d: (a) Tree of organisms expressing various forms of integrin-related proteins, from α-actinin to α-integrin includes diverse branches of single-celled eucaryotes (Sebe-Pedros et al. 2010). α-actinin was expressed in all branches. Both α- and β-integrin are expressed in those marked with (*) including single-celled Capsaspora owcarzaki and Amastigomonas sp. (b) Tree of tyrosine kinases (Suga et al. 2012). Cytoplasmic CTKs have a deep eucaryotic origin while receptor RTKs evolve later. (c) Tree of GPCR components extends back to LECA (Mendoza et al. 2014) making this signalling pathway, central to the nervous systems of higher animals, a founding unit of eucaryote evolution. All except those marked with * had one or more GPCR components and all clades included species possessing canonical GPCR, across both unikonta and bikonta, implying a common origin with LECA.
Examination of three key gene systems associated with the emergence of metazoa, the integrin pathway (Sebe-Pedros et al. 2010), tyrosine kinases (Suga et al. 2012) and G-protein coupled receptors (Mendoza et al. 2014)shows the key components evolved long before in single-celled eucaryotes and even the last eucaryote common ancestor in the case of G-protein system components. In both tyrosine kinases and GPCRs this allowed for a burst diversification of receptor-based tyrosine kinases and diverse GP-coupled receptors as the metazoa emerged. They have cell growth regulators such as p53, a gene notorious for cancer in humans. They have genes for cadherins and C-type lectins, proteins that help cells stick together, keeping a tissue intact. All told, by surveying the active genes in 21 choanoflagellate species, they have have been found to some 350 gene families once thought to be exclusive to multicellular animals.
Studies of Volvox, a spherical colonial alga, shows that multicellular organisms also found new ways to use existing functions. Whereas Volvox individuals have 500 to 60,000 cells arranged in a hollow sphere, some relatives, such as the Gonium species, have as few as four to 16 cells; others are completely unicellular. Volvox has repurposed other features of the single cell ancestor as well. In Chlamydomonas, an ancient stress response pathway blocks reproduction at night, when photosynthesis shuts down and resources are scarcer. But in Volvox, the same pathway is active all the time in its swimming cells, to keep their reproduction permanently at bay. Chlamydomonas under pressure of a population of paramecia that eat them and tend to pick off the smaller cells induced a kind of multicellularity was quick to appear: Within 750 generations, experimental populations had started to form and reproduce as groups.
In yeasts, cells selected for larger size began to form snowflake-like colonies. single mutation causes daughter cells to stick together. Cells specializing to die ealry also cause individual fronds to separate forming new colonies. Mutations in the cells released from each snow flake branch are passed on to all cells in the next colony. Consequently, subsequent snow flakes start out with new group traits.
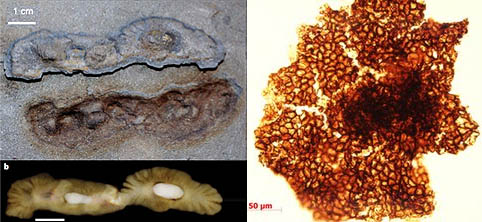

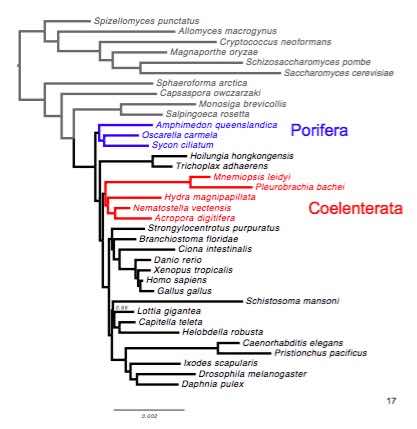 Fig 16e: Three fossils suggesting an early origin for multicellular life, Left: At 2.1 byrs (El Albani et al. 2010) Centre: At 1.5 byrs (Zhu et al. 2016). Near Right: 1.6 Bya old fossils resembling red algae found in Tirohan Dolomite of the Lower Vindhyan in central India (Bengtson et al. 2017)) Far Right Deep Homolog tree of animal origins (Pett et al. 2019).
Fig 16e: Three fossils suggesting an early origin for multicellular life, Left: At 2.1 byrs (El Albani et al. 2010) Centre: At 1.5 byrs (Zhu et al. 2016). Near Right: 1.6 Bya old fossils resembling red algae found in Tirohan Dolomite of the Lower Vindhyan in central India (Bengtson et al. 2017)) Far Right Deep Homolog tree of animal origins (Pett et al. 2019).
The view of the root of the metazoan tree has shifted significantly with the discovery of new genetic data, novel primitive species and new fossil evidence. Research into the absence of the miRNA pathway of ctenophores (Maxwell et al. 2012) and an apparently separate evolution of neural nets and apparent absence of HOX genes (Moroz et al. 2014) suggests that, rather than being sister organisms of the cnidaria, they may be more ancient life forms originating in the Ediacarian. Fossils of another organism Eoandromeda octobrachiata dating back to 580 million years (Tang et al. 2011) is consistent with this picture. The discovery of large colonial fossils dating back to 2.1 billion years (Albani et al. 2010) in addition to the previous discovery of the putative tubular alga Grypania spiralis dating back to a similar age (Han & Runnegar 1992) sets a potentially very early origin for multicellular organisms, also lending weight to the biological nature of Mawsonites spriggi (Seilacher et al. 2005).
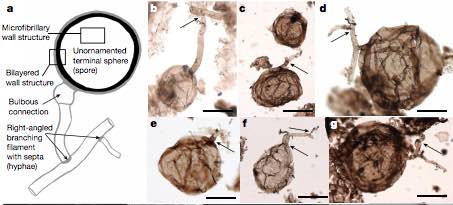


Fig 16e2: Left: Fungal fossil with hyphae dated to 0.9-1 Bya (Loron et al. 2019). Centre: The alga Proterocladus antiquus carpeted seafloor 1bn years ago and was size of rice grain.
Fossil plankton from half a billion years ago. An equivalent strategy is known only among green algae, specifically chlorophycean chlorophytes (Harvey 2023).
 To untangle the relationships among early-evolving animal groups, evolutionary biologists compare the sequences of 'homologous' genes from a range of species. Homologous genes have similar amino-acid sequences and code for proteins that often perform the same functions in the different species of interest and are therefore assumed to have been derived from a common ancestor. As a rule, these analyses have specifically focused on 'orthologs' - genes thought to be derived by vertical descent from a single ancestral sequence. But in the case of lineages that have undergone rapid diversification - those in which the rate of accumulation of novel mutations is especially high - genes can change so fast that it becomes difficult to confidently identify genes that are true orthologs. To avoid these drawbacks, Worheide and his colleagues (Pett et al. 2019) have adopted a different strategy, fig 16e far right, in which the presence or absence of homologous gene families rather than the comparison of orthologous gene sequences is the data of interest. This shift of viewpoint enables them to also take paralogs into account - products of the duplication of a pre-existing gene, which subsequently may evolve independently of each other. "When all homologous gene families are incorporated into a comparative phylogenetic analysis of gene content, there is a deeper store of information to draw upon.
To untangle the relationships among early-evolving animal groups, evolutionary biologists compare the sequences of 'homologous' genes from a range of species. Homologous genes have similar amino-acid sequences and code for proteins that often perform the same functions in the different species of interest and are therefore assumed to have been derived from a common ancestor. As a rule, these analyses have specifically focused on 'orthologs' - genes thought to be derived by vertical descent from a single ancestral sequence. But in the case of lineages that have undergone rapid diversification - those in which the rate of accumulation of novel mutations is especially high - genes can change so fast that it becomes difficult to confidently identify genes that are true orthologs. To avoid these drawbacks, Worheide and his colleagues (Pett et al. 2019) have adopted a different strategy, fig 16e far right, in which the presence or absence of homologous gene families rather than the comparison of orthologous gene sequences is the data of interest. This shift of viewpoint enables them to also take paralogs into account - products of the duplication of a pre-existing gene, which subsequently may evolve independently of each other. "When all homologous gene families are incorporated into a comparative phylogenetic analysis of gene content, there is a deeper store of information to draw upon.
Fig 16e3: Among the oldest known fossils of eukaryotic cells, estimated to be between 1.4 & 1.65 bya.
Above: A variety of fossils from the Limbunya Group in Northern Territory, Australia. Below: Tappania plana from Montana.
The discovery of a living genus Dendrogramma in Australia (Just et al. 2014) originally put it earlier than the emergence of the ctenophores, but later genetic evidence indicates this is a siphonophore, and hence in the cnidaria (Gough M 2016 Origin of mystery deep-sea mushroom revealed BBC 7 Jun).


Fig 16f: Left: Trees of eucaryotes based on translation elongation factor EF-2 and β-tubulin genes (King and Carroll). Right :Finding the root of the metazoan tree. (a) Ctenophora first tree elucidating fixation of identified components from miRNA to neurones and muscles (Moroz et al. 2014) (b) Evidence for early emergence of the Ctenphora exemplified by Mnemiopsis leidyi based on their lack of miRNA processing suggests they diverged before both sponges such as Amphimedon queenslandica and the placozoan Trichoplax adhaerens (Maxwell et al. 2012). (c) Possible evolutionary position of Dendrogramma (Just et al. 2014).
Looking at the varying pace of evolutionary change in a very ancient gene family and one of the largest in the human genome, the G-protein linked receptor family has roots going back to the first eucaryotes, with two major types of serotonin receptor 5HT1 and 5HT2 diverging before the molluscs, arthropods and vertebrates diverged and originating between 750 million and 1 billion yeas ago. Consequently serotonin functions in mood and circadin rhythms in a similar manner in insects and humans. The diversity of neurotransmitters in humans particularly the amines serotonin, dopamine, norepinephrine and histamine and the amino acids glutamate, gamma-amino-butyric acid and glycine originate from the need of single celled eucaryotes to communicate major pathways of strategic survival from nutrition through aversion to reproduction and sporulation. The family also includes the opsins of visual perception, development receptors and a diverse array of olfactory receptors which are evolving far more rapidly. Serotonin appears with the first photosynthetic bacteria that used tryptophan to hold the porphyrin reactive center and continues, along with melatonin to play a crucial role in light and circadian cycles in humans, as well as mood and social responsiveness.
See: Entheogens, the Conscious Brain and Existential Reality
 Fig 16g: (a) The two major serotonin receptor types 5HT1 and 5HT2 separated before the molluscs, arthropods and vertebrates diverged (Blenau & Thamm). (b) Evolutionary tree of the human G-protein linked receptors with examples highlighted in color. On the α branch are amine receptors - serotonin 5HT1A and 5HT2A, dopamine D1, and D2 (DRD1, DRD2), adrenergic α2a (ADRA2A), muscarinic acetylcholine (CHRM2), trace amine TAR1, as well as rhodopsin (RHO) and encephalopsin (OPM3). On the glutamate branch are metabotropic glutamate mGluR2 and GABA GABBR1. On the β branch is oxytocin (OXTR) surrounded by vasopressin receptors and Ghrelin. On the γ branch are opioid κ and μ (OPRK1, OPRM1).
Fig 16g: (a) The two major serotonin receptor types 5HT1 and 5HT2 separated before the molluscs, arthropods and vertebrates diverged (Blenau & Thamm). (b) Evolutionary tree of the human G-protein linked receptors with examples highlighted in color. On the α branch are amine receptors - serotonin 5HT1A and 5HT2A, dopamine D1, and D2 (DRD1, DRD2), adrenergic α2a (ADRA2A), muscarinic acetylcholine (CHRM2), trace amine TAR1, as well as rhodopsin (RHO) and encephalopsin (OPM3). On the glutamate branch are metabotropic glutamate mGluR2 and GABA GABBR1. On the β branch is oxytocin (OXTR) surrounded by vasopressin receptors and Ghrelin. On the γ branch are opioid κ and μ (OPRK1, OPRM1).  Olfactory and the non-rhodopsin receptors are linked to their respective points on the rhodopsin family tree. (Fredriksson R et al, Zozulya S. et al). (c) Insect tree of receptors for serotonin, dopamine, tyramine and octopamine neurotransmitters (Blenau & Baumann). Enlarged image right: Newly-discovered branch of bacterial heliorhodopsins (Pushkarev A et al. 2018). Microbial and animal visual rhodopsins(classified into type 1 and 2 rhodopsins correspondingly). Microbial rhodopsins are currently considered to be universal and the most abundant light harvesting proteins on Earth. Rhodopsins are present in all the three domains of life (bacteria, archaea and eukaryotes) as well as in giant viruses (Kovalev et al. 2019). Despite diversity of their functions and differences in the structures, all these rhodopsins are oriented in the membranes in the same way. Their N termini always face the outside of the cells. In 2018 a new large family of rhodopsins, named heliorhodopsins were discovered, facing the cytoplasmic space of the cell with their N termini. It was found that they are also present in Archaea, Bacteria, Eukarya and viruses.
Olfactory and the non-rhodopsin receptors are linked to their respective points on the rhodopsin family tree. (Fredriksson R et al, Zozulya S. et al). (c) Insect tree of receptors for serotonin, dopamine, tyramine and octopamine neurotransmitters (Blenau & Baumann). Enlarged image right: Newly-discovered branch of bacterial heliorhodopsins (Pushkarev A et al. 2018). Microbial and animal visual rhodopsins(classified into type 1 and 2 rhodopsins correspondingly). Microbial rhodopsins are currently considered to be universal and the most abundant light harvesting proteins on Earth. Rhodopsins are present in all the three domains of life (bacteria, archaea and eukaryotes) as well as in giant viruses (Kovalev et al. 2019). Despite diversity of their functions and differences in the structures, all these rhodopsins are oriented in the membranes in the same way. Their N termini always face the outside of the cells. In 2018 a new large family of rhodopsins, named heliorhodopsins were discovered, facing the cytoplasmic space of the cell with their N termini. It was found that they are also present in Archaea, Bacteria, Eukarya and viruses.
The evolution of the metazoan sodium channel essential for the neuronal action potential, from the Calcium channel shared by fungi and animals ocurred in single celled eucaryotes before the metazoa evolved from choanoflagellate-like ancestors. Metazoan eyes also appear to have a common origin, as indicated by the capacity of both jellyfish and mouse pax genes to elicit ectopic compound eyes on fruit files.
Fig 16h (a) Common involvement of PAX genes in eye formation from jellyfish, insects and vertebrates suggests a single common origin despite the differing mechanisms. The jelly fish pax genes, like mouse pax-6, induce ectopic compound eyes in fruit fly (right) (Suga et al, Kozmik et al). The small camera eyes of a jellyfish are shown top right (yellow arrow). It has even been suggested that the jellyfish could have gained the eye development pathway through symbiosis with certain single-celled dinoflagellates which possess an eyespot ocelloid, complete with lens and retinoid organelle (lower right) and may have in turn inherited this functionality from cyanobacterial chloroplasts via red algae (Pennisi, Keim, Le Page). Detailed analysis (Gavelis et al. 2015) shows it to be a compound endosymbitoic structure involving both a mitochondrial 'cornea' and red-alga plastid derived retinal body comprising stacked wave-form membranes derived from chloroplast thylakoids surrounded by pigmented lipid droplets. Dinoflagellates are thus an example of multiple engulfing endosymbiont events in which both mitochondria and then single-celled red-algae complete with plastids have been incorporated. (b) Evolutionary diversification of Na+ channels from Ca++ channels, essential for the action potential, appears to have occurred before the existence of nervous systems in founding single-celled eucaryotes leading to the metazoa before the choanoflagelates such as monosiga (Liebeskind et al).
Trees of Life based on Integrating Phylogenetic relationships and Environmental Genetic Diversity
There have been several recent efforts to construct comprehensive trees of life that include phylogenetic relationships that unite all lineages. To reconstruct a comprehensive tree of life, Hinchliff et al. (2015) synthesized published phylogenies, together with taxonomic classifications for taxa never incorporated into a phylogeny, presenting a draft tree containing 2.3 million tips - the Open Tree of Life.

Fig 16i: Phylogenies representing the tree. The depicted tree is limited to lineages containing at least 500 descendants. (A) Colors represent proportion of lineages represented in NCBI databases. (B) Colors represent the amount of diversity measured by number of descendant tips. (C) Dark lineages have at least one representative in an input source tree.

Fig 16j: Although the Open Tree of Life contains only one resolution at any given node, the underlying graph database contains conflict between trees and taxonomy, highlighting ongoing conflict near the base of Eukaryota (A) and Metazoa (B).
Following a complementary course, Hug et al. (2016) use new genomic data from over 1,000 uncultivated and little known organisms, using metagenomics - a shotgun sequencing-based method in which DNA isolated directly from the environment is sequenced, and the reconstructed genome fragments are assigned to draft genomes, together with published sequences, to infer a dramatically expanded version of the tree of life. The results reveal the dominance of bacterial diversification and underline the importance of organisms lacking isolated representatives, with substantial evolution concentrated in a major radiation of such organisms. This tree highlights major lineages currently underrepresented in biogeochemical models and identifies radiations that are probably important for future evolutionary analyses.


Fig 16k: (Left)Tree of life including uncharacterized species. (Right) Tree of life based on complete genome sequences
(Gringer commons.wikimedia.org/wiki/File:Tree_of_life_with_genome_size.svg).
A third comprehensive tree of life is based on completely sequenced genomes with genome sizes arrayed around the boundary.
Viral Influences on the Nuclear Genome

Fig 17: Viral eukaryogenesis
In viral eukaryogenesis (VE), (Bell PJ (2001, 2006, 2009, 2019, 2020) the nucleus arises form a DNA virus. The process started when a cell-wall less archaeon and an alpha-proteobacteria established a syntrophic relationship and evolved into asyntrophic consortium. A complex DNA virus then permanently lysogenized the host resulting in a tripartiteconsortium. The tripartite consortium consisted of the archaeon and the bacteria in a syntrophic relationship,and the archaeon and the virus in a host/parasite relationship. According to the VE hypothesis, the eukaryotic cytoplasm is descended from the original archaeal host, the mitochondria are descended from the bacterial endosymbionts, and the nucleus is descended from the complex DNA virus.1In the process of evolving from the tripartite consortium into a eukaryotic cell, genes from the alpha-proteobacteria and the archaeal host genome were transferred to the virus resulting in increased complexity of the virus, a reduction in the complexity of the alpha-bacterial genome and a complete loss of the independently replicating archaeal genome. In addition, during the process of eukaryogenesis, the three members of the consortium evolved to replicate as a coordinated single unit, and the process of phagocytosis evolved based on the membrane fusion proteins of the virus. The unique mode of replication of the unit as a whole was fundamentally different from any individual prokaryote or virus. A mode of replication where the virus was transmitted vertically through the archaeal host lineage led to the evolution of mitosis. A modified mode of replication where the virus induced conjugation of the host with other infected host consortia led to the evolution of sex and meiosis. This is supported by viral and eucaryote corresondences of key genes on the DNA replication pathwey and mRNA transcription. It is also supported bythe structure of large viruses such as mimi and medusa viruses (Takemura 2020), which include additional cellular genes providing greater control over the infection process (Chaikeeratisak et al. 2017a, b, Claverie 2006, Trevors 2003).
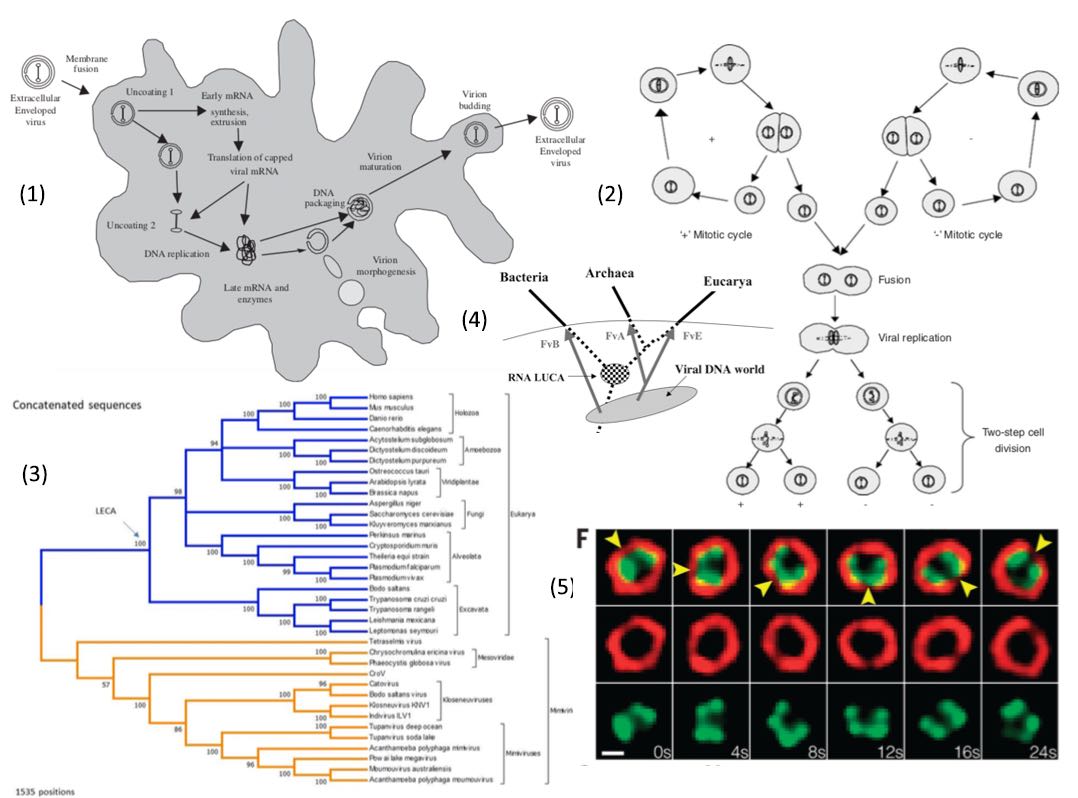
Fig 17a: (1) Infective process of a DNA virus can later take over control of the cell genetic machinery (2) Dual infection b closely related DNA viruses results in cell fusion and ultimately meiosis.(3) Correspondences between key genetic processes in genetic replication in viruses and eucaryotes. (4) Proposed viral contribution of DNA polymerases FvA etc. founder viruses to the three branches of life (Forterre). Other cellular and viral genealogies are possible and the scheme is merely representative.
Prokaryotes acquire genes from the environment via lateral gene transfer (LGT). Recombination of environmental DNA can prevent the accumulation of deleterious mutations. The greater complexity of eukaryotes is linked with larger genomes. Colnaghi, Lane, & Pomiankowski (2020) demonstrate that the benefit of LGT declines rapidly with genome size and that the degeneration of larger genomes can only be resisted by increases in recombination length, to the same order as genome size – as occurs in meiosis. It has been discovered that the protein system hapless 2/generative cell specific1 (HAP2/GCS1), promotes gamete fusion in organisms ranging from protists to flowering plants and insects, although it is strangely so far absent in vertebrates suggesting a newer mechanism has taken over. Key structural features of the HAP2 protein have been revealed, lending new insights into its mode of action and reinforcing its relationship to viral proteins that accomplish a similar task and may be intimately linked to the origins of cell–cell fusion events (including sexual reproduction) across evolutionary time (Clark 2018). The crystallographic structure determination of a candidate archaeal HAP2, FusexinA reveals an archetypical trimeric fusexin architecture with novel features such as a six-helix bundle and an additional globular domain. Moi D et al. (2021) demonstrate that ectopically expressed FusexinA can fuse mammalian cells, and that this process involves the additional domain and a more broadly conserved fusion loop. Genome content analyses reveal that archaeal fusexins genes are within integrated mobile elements, confirming a viral/TE origin.

Fig 17a2: HAP2
Forterre looks likewise to a three component origin, but his emphasis is on the idea that viruses have contributed major components to the genome of all three groups, possibly providing each of three RNA-based cell lineages with independent transitions to DNA-based genomes by contributing DNA-polymerases, thus radically improving the stability and competitiveness of these cell lines who became the eventual survivors. In addition to the ribosomal proteins and rRNAs having distinct qualitative features in each domain, many DNA informational proteins exist in different nonhomologous families (usually with several versions for one family). There are already six known nonhomologous families of cellular DNA polymerases. In the case of DNA polymerases of the B family, there is one version in Bacteria (only found in some proteobacteria), one in Archaea, and several in Eukarya. The distribution of the different versions and families of cellular DNA informational proteins among domains is erratic most of the time and does not fit with any of the models proposed for the universal tree, suggesting abrupt insertion into the cellular genomes by viral transfer.

Fig 17b: The differing DNA and RNA viral taxomomy of the Rep and capsid genes of BSL RHDV (Diemer & Stedman 2012).
A very unusual virus has given an indication how the transfer from RNA to DNA could have meen mediated by viruses. Although viruses are very promiscuous, they generally only recombine with viruses of s similar type or at least the same mode of replication. Thus until recently no instances were known of viral recombination bridging the three major groups: RNA viruses, DNA viruses and retroviruses encoding DNA from RNA instructions by reverse transcriptase. However a chimeric circular, putatively single-stranded DNA virus BSL RHDV encoding a major capsid protein similar to those found only in single-stranded RNA viruses was discovered in a hot acidic lake (Diemer & Stedman 2012). They also found that something very similar had turned up in samples of ocean water sequenced by a team led by Craig Venter. This gives the beginning of an explanation how DNA-based RNA-viral genes in endemic viruses, presumably via reverse transcriptase, have made multiple RNA to DNA transitions of other viral and cellular genes, probably when RNA, DNA and retroviruses cohabited cells.
Viruses such as phages appear to have no evolutionary tree, with genomes across widely diverse habitats consisting of cut and paste components, implying viral adaption has resulted almost entirely from utilization of advantageous genes from horizontal transfer. Around 10% of all bacterial genes sequenced to date consist of ORFans that bear no resemblance to genes seen anywhere else, suggesting horizontal viral origin (Hamilton).


Fig 18: Left: Evolutionary tree of DNA polymerase amino termini (Villareal and Defilippis)
Right: Bacterial DNA polymerases also show viral members (underlined) close to the root of the tree (File et. al.).
Villareal and Defilippis have likewise investigated the idea that DNA viruses are the origin of DNA replication proteins, by investigating the amino terminus and constructing an evolutionary tree which shows DNA polymerases of DNA viruses, eukaryotes (&alpha,&delta), archaea, E coli and two phages rooted in a tree consistent with a viral origin.
This idea has a great deal of plausibility because viruses are now know to have a potentially primal origin, rather than being recent escapees from cellular genomes which have undergone reductive parasitic changes to their genome. Viruses clearly also have retained both RNA-RNA, DNA-DNA and retrotranscription DNA-RNA-DNA using both RNA and DNA stages in their capsid viral forms, so they retain all the transitional states between RNA and DNA-based replication.
Furthermore the retroviruses and related mobile genetic elements have a common ancient evolutionary origin, which is related to telomerase, which itself uses an RNA primer to initiate chromosome duplication. There is thus a plausible case that telomerase is in fact a biological fossil of a retroviral conversion of the founding Eukaryote cell line to a DNA genome.
RNA viruses also have an ancient evolutionary relationship with their eucaryote hosts. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups (Koonin et al . 2008). More generally all the major groups of RNA viruses infecting humans and mammals, have been found to have an evolutionary origin going back to the origin of the vertebrates (Shi et al. 2018).
The Symbiotic Face of Eukaryote Mobile Elements

Fig 19: The deep relationship between transposable elements and chromosomal rearrangements in mammalian evolution. While humans have 23 chromosome pairs, dogs have 39 and some rodents 52. Left: Rearrangements of the original 21 pairs of ancestral eutherian chromosomes in reconstructed descendant ancestors, and extant descendant and outgroup species (Kim et al. 2017, Pennisi 2017). Harris Lewin, who led the study, thinks repetitive sections tend to make chromosomes susceptible to scrambling. Goats and cows, as well as rodents, have many retrotransposons, whereas primates have far fewer of both. Red blocks indicate eutherian chromosomes that were maintained, with shades of the color indicating the fraction of the chromosome affected by intrachromosomal rearrangements changing the order of the packaging of 22,000 vertebrate genes (lightest shade most affected). Split blocks were also affected by interchromosomal fissions and translocations. Shades of green indicate the fraction of an ancestral chromosome affected. BOR, boreoeutherian ancestor; CAT, catarrhini ancestor; EUA, euarchontoglires ancestor; EUT, eutherian ancestor; GAP, great apes ancestor; HUC, human–chimp ancestor; SIM, simian ancestor. As mammals evolved, early on, the rate at which chromosomes broke apart was stable, and relatively low, with eight per 10 million years. But 65 million years ago, the rate jumped, averaging 20 per 10 million years in primates other than the orangutan. So the orangutan chromosome setup looks the most like the ancient ancestor revealed by Kim's team, with eight ancient chromosomes intact. Humans have five such chromosomes and mice have just one. The researchers also showed that ancestral chromosome 20 is completely conserved in primates, but very much changed in goats and cows because of rearrangements within chromosomes. Rat chromosomes, too, are very different than the early eutherian's, due to inter-chormosomal events. Centre: Mouse and Human transposable element evolutionary history, including LINEs (yellow), SINES (light and dark blue), retrovirus-like LTR (long-terminal repeat) elements (green/cyan) and DNA transposons (red). This history extends back over the entire mammalian evolutionary epoch, with around 8% and 4.5% divergence respectively every 25 million years, indicating the very ancient basis of this relationship, which extends 105 million years to the eutherian radiation (Human Genome Consortium, Waterston et. al. 2002). The most notable difference between human and mouse is in the changing rates of transposition over time. The rate has remained fairly constant in mouse,but markedly increased to a peak at about 40 Myr in human, and then plummeted. Short-lived species like the mouse can sustain a higher rate of transposition due to their shorter generations times. Beyond this overall tendency, there are specific differences in each of the four repeat classes. The extant L1 elements in both species derive from a common ancestor. L1 seems to have remained highly active in mouse, whereas it has declined in the human lineage. Whereas only a single SINE (Alu) was active in the human lineage, the mouse lineage has been exposed to four distinct SINEs (B1, B2, ID, B4). Each is thought to rely on L1 for retroposition. The mouse B1 and human Alu SINEs are unique among known SINEs in being derived from 7SL RNA; they probably have a common origin. The mouse B2 is typical among SINEs in having a transfer RNA-derived promoter region. Recent ID elements seem to be derived from a neuronally expressed RNA gene called BC1, which may itself have been recruited from an earlier SINE. MIR died out when the older line-L2on which it depended became extinct in both lineages. All interspersed LTR- containing elements in mammals are derivatives of the vertebrate-specific retrovirus clade of retrotransposons. Endogenous retroviruses fall into three classes, which show a markedly dissimilar evolutionary history in human and mouse. Class 3 accounts for 80% of recognized LTR element copies predating the human-mouse speciation. Notably, ERVs are nearly extinct in human whereas all three classes have active members in mouse. Copies of class 2 elements are tenfold denser in mouse than in human. In contrast, class I element copies are fourfold more common in the human genome. The fourth repeat class is the DNA transposons. Although most transposable elements have been more active in mouse than human, DNA transposons show the reverse pattern. Right: Evolutionary diversification of LINE elements spans plants and diverse animal phyla (Ivancevic et al. 2016) and extends also to amoebozoa (Malicki et al. 2017) indicating an origin concommitant with LECA.
In 1978, following the work of Darryl Reanny (1974-6), I proposed (1978, 1992) that viruses and transposable elements, far from just being selfish genes (Dawkins 1976), formed part of a dynamical system of genetic symbiosis between the hosts and the mobile genetic elements, because the mobile elements permitted forms of coordinated gene expression and the formation of new genes in a modular manner, which would otherwise be impossible, achieving in return perpetuation of their own genomes over evolutionary time scales. Most of the details of this proposal have proved to be realized. The ENCODE project has demonstrated involvement of all the major classes of human transposable element in regulatory enchancer activity, most specific to a single cell type (Thurman 2012).

![]()
Fig 20: Left: ENCODE data showing involvement of the major classes of transposable element in enhancer activity (Thurman 2012).
Right:
L1 replication: L1 is transcribed in open reading frames ORF1, an RNA-binding protein, and ORF2 an endonuclease/reverse-transcriptase.
The bound RNA-protein complex RNP is transported to the nucleus where target-primed reverse transcription to chromosomal DNA takes place (Han and Boeke).
By some reckonings, 40 to 50 per cent of the human genome consists of DNA imported horizontally by viruses, some of which has taken on vital biological functions. Taken together, virus-like genes represent a staggering 90 per cent of the human genome (Hamilton). Coding sequences comprise less than 5% of the human genome, whereas repeat sequences account for at least 50% and probably much more. Transposable LINE or long-intermediate repeat retroelements, common to mammals (Han and Boeke), and insects (Jensen et al, Sheen et al) with a history running back to the Eukaryote origin are specifically activated in both sperms and eggs during meiosis (Branciforte and Martin, Tchénio et. al., Trelogan and Martin), although subjected to down regulation by interfering piRNAs (Aravin et al). They replicate from transcribed RNA copies of themselves thus using RNA to instruct DNA copies, indicating an origin in RNA-based life, as does the active RNA processing of our own Eukaryote cells. Their RNA-based reverse transcriptase shows homologies with the telomerase essential for maintaining immortality in our germ line, indicating a common and symbiotic origin. 100,000-950,000 partially defective LINEs, around 100 of which remain fully active in humans, and their 300,000 dependent smaller fellow traveller Alu SINEs make up a significant portion of the human and mammalian genomes, along with pseudogenes, apparently defective copies of existing genes translocated by elements such as LINEs.
These elements travel passively down the germ line with chromosomal DNA, so their specific activation during meiosis suggests they may perform a role of coordinated regulatory mutation. This suggests that the type of symbiotic sexuality embraced by bacteria and plasmids also continues to function in higher organisms in a form of sexual symbiosis between our chromosomes and transposable genetic elements. This is consistent with the 1.4% point mutation divergence between humans and chimps, being overshadowed by an additional 3.9% divergence to 5.4% overall (Britten), when insertions and deletions are accounted.
SINEs, such as human Alu, a free-rider on the LINE reverse transcriptase derived from the small cellular RNA used to insert nascent proteins through the membrane, are in turn implicated in active functional genes (Reynolds, Schmid) particularly some involved in cellular stress reactions, again suggesting genetic symbiosis. Humans have about 13 times as many RNA edits as non-primate species, including inosine insertions associated with Alu elements, as well as intron deletions (Holmes ) and newly inserted exons (Ast), which may differentiate humans from other apes through alternative splicing of genes expressed in the brain. RNA editing is abundant in brain tissue, where editing defects have been linked to depression, epilepsy and motor neuron disease. There is a new Alu insert about every 100 births. As many as three quarters of all human genes are subject to alternative splice editing.
Somatic variation in LINE L1 insertions has been found in the human brain (Erwin et al. 2016, Singer et al. 2010). The healthy human brain is a mosaic of varied genomes. L1 retrotransposition is known to create mosaicism by inserting L1 sequences into new locations of somatic cell genomes. Somatic L1-associated variants in the brain (SLAVs) are composed of: (a) L1 retrotransposition insertions and (b) retrotransposition-independent L1-associated variants. A subset of SLAVs comprise somatic deletions generated by L1 endonuclease cutting activity. Retrotransposition-independent rearrangements in inherited L1s resulted in the deletion of proximal genomic regions. These rearrangements were resolved by microhomology- mediated repair, suggesting L1-associated genomic regions are hotspots for somatic copy number variants in the brain and therefore a heritable genetic contributor to somatic mosaicism. SLAVs are present in crucial neural genes, such as DLG2 and affect 44–63% of the cells in the healthy brain. Some transposon hopping may be a reaction to stress that may allow brain cells to develop capabilities not initially encoded in the genome which could influence behavior, thinking and personality. Even identical twins may have genetically different brain cells because of transposon hopping after the embryo splits.
Recent explosion of the area of interfering miRNAs as regulatory elements in gametogenesis and development (Großhans) has provided an explanation of how pseudogenes, including those retrotransposed via LINE elements, can gain functional regulatory significance even though they do not produce translatable mRNAs.
Centromeres, the anchor regions where microtubules from the centriole link to the chromosome, essential for chromosome division, have been found to contain selfish DNA that attempts to propagate itself exclusively during meiosis, when the division in females is asymmetric, with three of the four duaghter cells becoming polar bodies rather than an ovum, and thus do not get propagated to the next generation. Centromeres differ in their binding strength and stronger ones can detect their orientation within the dividing cell due to proteins released by the membrane to induce asymmetric division. When they find themselves in the region leading to a polar body, they repeatedly let go, focing the opposing centromere to do likewise, until due to random movements within the cell, they find themselves on the ovum pole and then draw tight ensuring their transmission along with the chromatids that contain them in the ovum. These centromeres appear to be genuinely selfish because centromere-binding proteins have been found to be one of the most rapidly evolving genes in the human genome, implying that they are caught in an arms race of mutually-antagoistic co-evolution (Akera T et al. 2017).

 Fig 21: Left: Pseudogene-mediated production of endogenous small interfering RNAs (endo-siRNAs). Pseudogenes can arise through the copying of a parent gene (by duplication or by retrotransposition). (a) An antisense transcript of the pseudogene and an mRNA transcript of its parent gene can then form a double-stranded RNA. (b) Pseudogenic endo-siRNAs can also arise through copying of the parent gene as in a and then nearby duplication and inversion of this copy. The subsequent transcription of both copies results in a long RNA, which folds into a hairpin, as one half of it is complementary to its other half. In both a and b, the double-stranded RNA is cut by Dicer into 21-nucleotide endo-siRNAs, which are guided by the RISC complex to interact with, and degrade, the parent gene's remaining mRNA transcripts. The mRNA from genes is in red and that from pseudogenes is in blue. Green arrows indicate DNA rearrangements (Sasidharan and Gerstein). Right: LINE-1 is essential for progression from the 2-cell embyo to the blastula in mice.
Fig 21: Left: Pseudogene-mediated production of endogenous small interfering RNAs (endo-siRNAs). Pseudogenes can arise through the copying of a parent gene (by duplication or by retrotransposition). (a) An antisense transcript of the pseudogene and an mRNA transcript of its parent gene can then form a double-stranded RNA. (b) Pseudogenic endo-siRNAs can also arise through copying of the parent gene as in a and then nearby duplication and inversion of this copy. The subsequent transcription of both copies results in a long RNA, which folds into a hairpin, as one half of it is complementary to its other half. In both a and b, the double-stranded RNA is cut by Dicer into 21-nucleotide endo-siRNAs, which are guided by the RISC complex to interact with, and degrade, the parent gene's remaining mRNA transcripts. The mRNA from genes is in red and that from pseudogenes is in blue. Green arrows indicate DNA rearrangements (Sasidharan and Gerstein). Right: LINE-1 is essential for progression from the 2-cell embyo to the blastula in mice.
L1 elements have also been found to replicate in neural progenitor cells in both the mouse and human and copy numbers have been found to increase in the hippocampus, and in several regions of adult human brains, when compared to the copy number of endogenous L1s in heart or liver genomic DNAs from the same donor. The authors comment that these data suggest that de novo L1 retrotransposition events may occur in the human brain and, in principle, have the potential to contribute to individual somatic mosaicism (Coufal et. al. 2009 L1 retrotransposition in human neural progenitor cells Nature doi:10.1038/nature08248).
L1 is paradoxically highly expressed during early development and plays essential roles in mouse embryonic stem cells (ESCs) and pre-implantation embryos (fig 21). In ESCs, LINE-1 acts as a nuclear RNA scaffold that recruits Nucleolin and Kap1/Trim28 to repress Dux, the master activator of a transcriptional program specific to the 2-cell embryo. In parallel, LINE-1 RNA mediates binding of Nucleolin and Kap1 to rDNA, promoting rRNA synthesis and ESC self-renewal. In embryos, LINE-1 RNA is required for Dux silencing, synthesis of rRNA, and exit from the 2-cell stage. The results reveal an essential partnership between LINE-1 RNA, Nucleolin, Kap1, and peri-nucleolar chromatin in the regulation of transcription, developmental potency, and ESC self-renewal (Percharde et al. 2018).
Although the data from the human genome project indicated that human LINEs are becoming less active as a group by comparison with the corresponding elements in the more rapidly evolving mouse genome, there remain about 60 active human LINE elements which are known to be responsible for mutations in humans. More recent investigation (Boissinot et. al.) shows that the most recent families are highly active. Around four million years ago shortly after the chimp-human split, a new family Ta-L1 LINE-1 emerged and is still active, with about half the Ta insertions being polymorphic, varying across human populations. Moreover 90% of Ta-1d, the most recent subfamily are polymorphic, showing highly active lines remain present. LINEs are more heavily distributed on the sex chromosomes with X chromosomes containing 3 times as many full length potentially active elements and the Y chromosome 9 times as many! This is consistent with a continuing mutational load on humans which is removed more slowly from the sex chromosomes by crossing over in proportion to the degree to which crossing over is inhibited in each (i.e. totally on the Y and largely in males in the X but not in females). Sexual recombination is a protection from mutational error in a process called Muller's ratchet.
 Fig 22: Evolution of reverse transcriptases from a common ancestor bearing a LINE archetype (Xiong and Eickbush, Nakamura et. al.). The root of their evolution goes back to the transfer from RNA to DNA at the beginning of life. They form a complementary evolutionary tree to that of cellular life as genetic symbionts of metazoa travelling down the germ line. Their group includes telomerases essential to the reproductive cycle.
Fig 22: Evolution of reverse transcriptases from a common ancestor bearing a LINE archetype (Xiong and Eickbush, Nakamura et. al.). The root of their evolution goes back to the transfer from RNA to DNA at the beginning of life. They form a complementary evolutionary tree to that of cellular life as genetic symbionts of metazoa travelling down the germ line. Their group includes telomerases essential to the reproductive cycle.
LINEs are preferentially expressed in both steriodogenic and germ-line tissues in mice (Branciforte and Martin, Trelogan and Martin), suggesting stress could interact with meiosis. L1 expression occurs in embryogenesis, at several stages of spermatogenesis including leptotene, and in the primary oocytes of females poised at prophase 1. This could enable somatic stress to have a potential effect on translocation in the germ-line which might enable form of genetic adaption in long-lived species such as humans. Conversely the SRY-group male determining gene SOX has been found to regulate LINE retrotransposition (Tchénio et. al.). Similarly LINE elements have been found to be 'boosters' in the inactivation of one X chromosome that happens in female embryogenesis (Lyon 2000, Chow et al. 2010).
LINE-1 retrotransposons, one of the main components of heterochromatin, are also highly transcribed in the first activation of maternal and paternal genes in the zygote. In most of our cells these transposons are silent, and so most researchers had considered retrotransposon activation to be a side effect of the overall reprogramming process. But when transcription activator-like effectors (TALEs), were used to prevent LINE-1 activation, decreased rates of development resulted. However, adding LINE-1 mRNAs to make up for the lack of transcription did not rescue the phenotype, so it's not the messenger RNA itself, but just what is happening the DNA loci, suggesting that retrotransposon activation initiates zygotic gene expression, helping open up the chromatin, so that other elements that direct transcription in other genes can function more efficiently (Akst J 2017 New Techniques Detail Embryos' First Hours and Days The Scientist 1 Dec).
They have diverse means both to cause mutational damage and novel alleles (Han and Boeke).
Both L1 and Alu elements may be able to self-regulate rates of replication, through the existence of stealth drivers, viable elements which maintain a low transcription rate of active elements, with little genomic impact and hence little negative selection. These occasionally seed daughter master elements, which may replicate actively to form new families when conditions permit. This picture is consistent with long periods of quiescence, punctuated by bursts of 'saltatory' replication leading to large copy numbers (Han et. al.). These are the only TEs unequivocally shown to be currently active in humans, as demonstrated by de novo insertions causing genetic disorders (Cordaux & Batzer 2009), including loss of the tail in apes (Xia et al. 2024).
 In both mice and humans, the placenta utilises SINE elements (Alu and B1) to form ds-RNA to modulate the immune system in pregnancy to induce type III interferon to avoid viral disease while not inducing rejection of the embryo(s). By pretending it's under viral attack, it keeps the immune system running at a gentle, steady pace to protect the enclosed foetus from viruses that slip past the mother's immune defences (Wickramage et al. 2023).
In both mice and humans, the placenta utilises SINE elements (Alu and B1) to form ds-RNA to modulate the immune system in pregnancy to induce type III interferon to avoid viral disease while not inducing rejection of the embryo(s). By pretending it's under viral attack, it keeps the immune system running at a gentle, steady pace to protect the enclosed foetus from viruses that slip past the mother's immune defences (Wickramage et al. 2023).
Further evidence of a symbiotic relationship comes from Drosophila telomeres, which are maintained by the non-LTR retrotransposons, the Line-like TART (Jensen et al, Sheen et al) and HeT-A (Biessmann et al). Likewise the recombination activating gene protein RAG1/RAG2, essential for the mutational variability of the vertebrate immune system, appears to have evolved from an ancient DNA transposon common to the metazoa (Agrawal et al, Kapitinov & Jurga 2005). Significant similarities exist in the catalytic proteins of Hermes hAT transposase in insects, the V(D)J recombinase RAG, and retroviral integrase superfamily transposases, thereby linking the movement of transposable elements and V(D)J recombination (Zhou et al).
![]() Fig 22b: Transib evolutionary tree spans the eucaryotes (Kapitonov and Jurka).
Fig 22b: Transib evolutionary tree spans the eucaryotes (Kapitonov and Jurka).
Recombination-activating genes (RAGs) encode enzymes that play an important role in the rearrangement and recombination of the genes of immunoglobulin and T cell receptor molecules. Researchers had long suspected that the two DNA-cutting enzymes RAG1 and RAG2 are encoded by relics of a DNA transposon, but no one had ever found a transposon that uses those proteins. Working with the primordial vertebrates called lancelets, Xu and colleagues found a DNA transposon called ProtoRAG implying the DNA transposon that gave rise to the two enzymes jumped into an ancestor of lancelets and jawed vertebrates about 550 million years ago (Saey 2017). The approximately 600-amino acid ‘‘core’’ region of RAG1 required for its catalytic activity is also significantly similar to the transposase encoded by DNA transposons of the Transib superfamily discovered recently based on computational analysis of the fruit fly and African malaria mosquito genomes. Transib transposons also are present in the genomes of sea urchin, yellow fever mosquito, silkworm, dog hookworm, hydra, and soybean rust (Kapitonov and Jurka).
Transposable elements have major evolutionary influences on higher eucaryote evolution, in ways which make them central to phenotypic complexity and identity (Saey 2011, 2017). As entities that make their living by getting copied into RNA over and over again, retrotransposons are littered with transcription factor binding sites. Gene modules such as those arising from the bounding long terminal repeats or LTRs of transposable elements can disseminate whole libraries of binding sites that over time become complex gene-regulating switches.
Some of these recycled transposon factors may have helped humans fight viruses. About 45 million to 60 million years ago, a retrovirus called MER41 invaded the genome of a primate ancestor of humans. MER41 includes binding sites for transcription factors involved in fighting infections which are alerted to infection by interferon gamma. The retrovirus may have used the interferon gamma signal to boost its own production. But over time, the mammalian hosts turned that weapon against the virus.
Rewiring gene activity in humans happened, in part, when transposons inserted themselves into the genomes of human ancestors after the split from chimpanzees. Remains of the transposons that infected humans have been recycled into more than a thousand regulatory switches found only in humans.
 Fig 22b1: Arc retrovirus like capsids (right) and the evolutionary relationships of fly and tetrapod Arc genes with retrotrnsposon.
Fig 22b1: Arc retrovirus like capsids (right) and the evolutionary relationships of fly and tetrapod Arc genes with retrotrnsposon.
A key brain gene called Arc has been recently discovered to be an ancient version of the gag gene essential to retrotrasposons and retroviruses, related in particular to the gypsy retrotransposon in fruit flies indicating that it is ancient and not derived from retroviruses themselves. The neuronal gene Arc is essential for long-lasting information storage in the mammalian brain, mediates various forms of synaptic plasticity, and has been implicated in neuro-developmental disorders. Endogenous Arc protein is released from neurons in extracellular vesicles that mediate the transfer of Arc mRNA into new target cells, where it can undergo activity-dependent translation. A mouse that's born without Arc can't learn or form new long-term memories. If it finds some cheese in a maze, it will have completely forgotten the right route the next day. Arc is key to transducing the information from experiences into changes in the brain. Arc is capable of both endocytosis – drawing in receptors from the synaptic membrane – and packing and transmitting m-RNAs in viral budding corpuscles to other neurons. Fruit flies have Arc genes that descend from the same group of gypsy retrotransposons that gave rise to ours. Shepherd and colleagues estimate that 350-400 million years ago, the retrotransposon, entered a land-based, tetrapod. This led to the development of the Arc protein, as it operates in our neurochemistry today. According to an earlier study, the same process developed in fruit fries, independently, around 150 million years ago. And yet, the fly versions of Arc also sens RNA between neurons in virus-like capsules (Pastuzyn et al. 2018).
MicroRNAs (miRNAs) are crucial regulators of gene expression at the post-transcriptional level in eukaryotes by targeting gene 3'-untranslated regions. Researchers identified 409 TE-derived miRNAs, 386 of which overlapped with TEs, which are derived from TEs in human, indicating that TEs play important roles in origin of human miRNAs, with humans also having more than other mammals and vertebrates.
![]() Fig 22b2: Gene birth by transposase capture in tetrapods. By inserting functional domains into new genomic contexts, transposase sequences can generate host-transposase fusion (HTF) genes through alternative splicing. Several genes with critical developmental functions, such the Pax transcription factors key to eye development, are thought to have been born through this process. Tetrapod phylogenetic tree with boxes representing HTF fusion genes. Colors indicate the transposase superfamily assimilated. Numbers in parentheses indicate the number of HTF genes identified in the specified lineage. OWM, Old World monkeys; NWM, New World monkeys.s.
Fig 22b2: Gene birth by transposase capture in tetrapods. By inserting functional domains into new genomic contexts, transposase sequences can generate host-transposase fusion (HTF) genes through alternative splicing. Several genes with critical developmental functions, such the Pax transcription factors key to eye development, are thought to have been born through this process. Tetrapod phylogenetic tree with boxes representing HTF fusion genes. Colors indicate the transposase superfamily assimilated. Numbers in parentheses indicate the number of HTF genes identified in the specified lineage. OWM, Old World monkeys; NWM, New World monkeys.s.
Cosby et al. (2021) confirm that exon shuffling is a major evolutionary force generating genetic novelty. We provide evidence that DNA transposons promote exon shuffling by inserting transposase domains in new genomic contexts. This process provides a plausible path for the emergence of several ancient transcription factors with important developmental functions.
Into the transgenetic milieu also come lincRNAs - non-coding RNAs over 200 nucleotides in length that are generally but not necessarily inter-genic have reamined enigmatic but are increasingly associated with key protein traffic management roles in the eucaryote cell, serving as guides showing proteins where to go, tethering proteins to different types of RNA, or to DNA, acting as decoys and distracting regulatory molecules from their usual assignments, moulding cellular development and the phenotypic features of every organismic species, which are defined by regulatory variations in a largely similar complement of protein-oding genes. Although genes that code for proteins make up only 1.5% of the mouse genome, more than 63% of the genome's DNA is copied into RNA. In humans the number is even higher, with up to 93% of the genome made into RNA, even though protein-coding genes make up less than 2% of the genome. According to experimental estimates there are 6,736 - 10,000 long noncoding RNAs encoded in the human genome, a figure comparable to the 20,000 protein coding genes. More than 30 percent of long noncoding RNAs are repurposed transposable elements. Humans have several lincRNAs that are found in no other species. Many of those RNAs are made in the brain, leading scientists to speculate that the molecules may be at least partially responsible for that important organ's evolution.
Examples are XIST and it nemesis TSIX which produce competing RNAs that inactivate one of the two X-chromosomes in females by coating one X in XIST. XIST has also been found to interact with LINE-1 elements to ensure X-inactivation doesn't escape repression, providing a key role for the elements in the life cycle (Lyon 2000, Chow et al. 2010).
Another is HOTAIR which is functionally involved in 854 distinct locations and inactivates HOXD developmenal genes also involved in cancer and it complementing activator HOTTIP. A computerized search of the human genome has identified 7,000 genes whose m-RNAs could act as microRNA decoys in 248,000 interactions. A pseudogene (a an inactive mutated version of a gene originating from a transposable element) has been confirmed to act as a decoy attracting microRNAs that bind to and inactivate m-RNAs for important genes such as PTEN involved in cancer suppression. the linc-RNA linc-MD1 is important in muscle development. In sponges two microRNAs away from the messenger RNA of two genes, allowing more muscle-building proteins to be made from those genes.
The competition between the tendency of retroelements to replicate explosively in the germ line and cellular control is maintained through the RNA silencing effects of PIWI-interacting small RNAs (piRNAs) and various nuclear and cytoplasmic accessories using RNA interference (Zamudio & Bourchis 2010). This mechanism appears to have emerged right at the origin of LECA because modular components of the key proteins have been identified as coming from two sources (a) a phage in the early proteobacterial endosymbiont contributing both an RNA dependent RNA polymerase and RNAaseIII and (b) the founding archaeum contributing an argonaute and a helicase (Shabalina & Koonin 2008), leading to the radiation of the composite proteins PIWI, Ago and Dicer. Archaea are also known to contain argonautes related to the eucaryote versions (Swarts et al. 2014). Piwi has been proposed to function with downstream partners, and one of them is the heterochromatin Protein 1a (HP1a), which reportedly enforces transposon silencing in the Drosophila germline and ovarian somatic cells (Teo et al. 2018).
Piwi-interacting RNA (piRNA) is the largest class of small non-coding RNA molecules expressed in animal cells. piRNAs form RNA-protein complexes through interactions with piwi proteins. piRNAs direct the piwi proteins to their transposon targets. These piRNA complexes have been linked to both epigenetic and post-transcriptional gene silencing of retrotransposons and other genetic elements in germ line cells, particularly those in spermatogenesis. The majority of piRNAs are antisense to transposon sequences, suggesting that transposons are the piRNA target. In mammals it appears that the activity of piRNAs in transposon silencing is most important during the development of the embryo, and in both C. elegans and humans, piRNAs are necessary for spermatogenesis. Three piwi subfamily proteins - MIWI, MIWI2 and MILI - have been found to be essential for spermatogenesis in mice. A decrease or absence of PIWI gene expression is correlated with an increased expression of transposons and a reduction in fertility. piRNA and endogenous small interfering RNA (endo-siRNA) may have comparable and even redundant functionality in transposon control in mammalian oocytes.
 Fig 22b3: Ping-pong RNA-cleavage between Fem and Masc in the silk worm. Birds, some fishes, butterflies and moths have a reverse female-determining chromosome system with ZW being female and ZZ being male. In the silkworm, the W chromosome is almost fully occupied with transposable element sequences and seems to have no protein producing genes at all, while a small PIWI-interacting RNA (piRNA) Fem, which silences an opposing DNA-binding zinc-finger protein of the male gene Masc on the Z by m-RNA cleavage, appears to determine sex (Kiuchi T. et al. 2014).
Fig 22b3: Ping-pong RNA-cleavage between Fem and Masc in the silk worm. Birds, some fishes, butterflies and moths have a reverse female-determining chromosome system with ZW being female and ZZ being male. In the silkworm, the W chromosome is almost fully occupied with transposable element sequences and seems to have no protein producing genes at all, while a small PIWI-interacting RNA (piRNA) Fem, which silences an opposing DNA-binding zinc-finger protein of the male gene Masc on the Z by m-RNA cleavage, appears to determine sex (Kiuchi T. et al. 2014).
The biogenesis of piRNAs is not yet fully understood, although possible mechanisms have been proposed. A primary processing pathway is suggested to be the only pathway used to produce pachytene piRNAs; in this mechanism, piRNA precursors are transcribed resulting in piRNAs with a tendency to target 5' uridines. The Ping Pong mechanism wherein primary piRNAs recognise their complementary targets and cause the recruitment of piwi proteins. This results in the cleavage of the transcript at a point ten nucleotides from the 5' end of the primary piRNA, producing the secondary piRNA. These secondary piRNAs are targeted toward sequences that possess an adenine at the tenth position. Since the piRNA involved in the ping pong cycle directs its attacks on transposon transcripts, the ping pong cycle acts only at the level of transcription. In silk worms, PIWI ping-pong amplification is involved in sex determination (Kiuchi et al. 2014). piRNAs can be transmitted maternally, and based on research in D. melanogaster, piRNAs may be involved in maternally derived epigenetic effects. Ping-pong signatures have been identified in very primitive animals such as sponges and cnidarians, pointing to the existence of the ping-pong cycle already in the early branches of metazoans.
The piRNA Ping-Pong pathway was first proposed from studies in Drosophila where the piRNA associated with the two cytoplasmic Piwi proteins, Aubergine (Aub) and Argonaute-3 (Ago3) exhibited a high frequency of sequence complementarity over exactly 10 nucleotides at their 5' ends.This relationship is known as the "ping-pong signature" and is also observed in associated piRNA from Mili and Miwi2 proteins isolated from mouse testes. The proposed function of Ping-Pong in Drosophila or in mouse remains to be understood, but a leading hypothesis is that the interaction between Aub and Ago3 allows for a cyclic refinement of piRNA that are best suited to target active transposon sequences.

Fig 22b4: Top row: Human Blastocyst expression of HERVK. DAPI is a flourescent dye binding to AT-rich regions, OCT4 is a key embroygenesis stem factor (Grow et al. 2015):
Bottom: Left, Graduated involvement of various linc-RNAs in embryogenic differentiaton and right, XIST binding to one of the two X's (Saey 2011).
Endogenous Retroviruses, Embryogenesis and the Placenta
Retroviral DNA - remnants of ancient retrovirus infections of germline cells - comprises 8% of the modern human genome. They are one of the oldest viral groups with evidence for an origin >450 mya ago with the first marine vertebrates or earlier (see fig 22b2). Endogenous retroviruses, or ERVs, which also travel down the germ line as free-riders, although some may retain infectious capacity, may be essential for placental function, as every mammal tested has placental blooms of endogenous retroviruses which appear to both aid the formation of the syncytium, the super-cellular fused membrane that enables diffusion from the mother to the baby and the immunity suppression, which prevents rejection of the embryo, both characteristics of retroviruses such as HIV.
Pluripotent stem cells are capable of generating all embryonic cell lineages but, until recently, scientists could seldom manipulate induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) to generate extra-embryonic cell types, such as placental cells. Prior work had shown that a small number of cells independently develop the potential to produce extra-embryonic cell types, and that the process was linked to endogenous retroviruses. Choi et. al (2017) have now shown that removing microRNA miR-34a from a stem cell can kick off a molecular pathway that induces endogenous retroviruses and, at the same time, enables iPSCs and ESCs to consistently form extra-embryonic cells in a dish. The results suggest that a particular class of noncoding RNA works in concert with the latent viral elements of the genome work to limit stem cell potential, and that removing a key miRNA can lift this limitation.
Brattas et al. (2017) have determined that almost 10,000, primarily primate-specific, ERVs may serve as "docking platforms" for a protein called TRIM28. Two years ago, Johan Jakobsson's team showed that ERV have a specific regulatory role in mouse neurons specifically. However, their 2017 study has been made using human cells. TRIM28 has the ability to "switch off" not only viruses but also the standard genes adjacent to them in the DNA helix, allowing the presence of ERV to affect gene expression. These results uncover a gene  regulatory network based on ERVs that participates in control of gene expression of protein-coding transcripts important for brain development. This switching-off mechanism may also behave differently in different people, since retroviruses are a type of potentially transposable genetic material that may end up in different places in the genome. This makes it a possible tool for evolution, and even a possible underlying cause of neurological diseases. There are further studies that indicate a deviating regulation of ERV in several neurological diseases such as ALS, schizophrenia and bipolar disorder.
regulatory network based on ERVs that participates in control of gene expression of protein-coding transcripts important for brain development. This switching-off mechanism may also behave differently in different people, since retroviruses are a type of potentially transposable genetic material that may end up in different places in the genome. This makes it a possible tool for evolution, and even a possible underlying cause of neurological diseases. There are further studies that indicate a deviating regulation of ERV in several neurological diseases such as ALS, schizophrenia and bipolar disorder.
Fig 22b5: Left evolution of functional TRIM28-binding ERVs with full length ERVs in red.
Right: Deelopmental expression of ERVs.
When the team analyzed ERV expression in detail, they found distinct differences between pluripotent human embryonic stem cells, which correspond to a developmental stage before germline commitment, and samples obtained from human embryonic brain. Although the majority of reads in neural cells originate from non-internal ERV fragments, hESCs transcribe a large number of internal ERV fragments, indicating that more complete ERV loci are primarily expressed in hESC, whereas long terminal repeat (LTR) fragments dominate in embryonic brain samples. Because the majority of ERVs expressed during human brain development were incomplete fragments, these loci may primarily be passively ex- pressed due to their position in a transcriptionally active genomic region.
Only one retrovirus, the most recent endogenous retrovirus to infect the human line, the HML2 subgroup of Human Endogenous Retrovirus K (HERV-K), has continued to reinfect the human line after the divergence from the lineage leading to chimpanzees and bonobos approximately 6 million years ago. HERV-K has remained active since then, reinfecting germ lineage cells of Neanderthals and Denisovans multiple times, around the time of, or subsequent to, the divergence of the archaic hominin lineages from that leading to modern humans (Agoni et al. 2012, Lee et al. 2014). One of the proviruses was shared by Neanderthals and Denisovans, consistent with these archaic humans sharing a common ancestor more recently than they shared one with the lineage leading to modern humans.
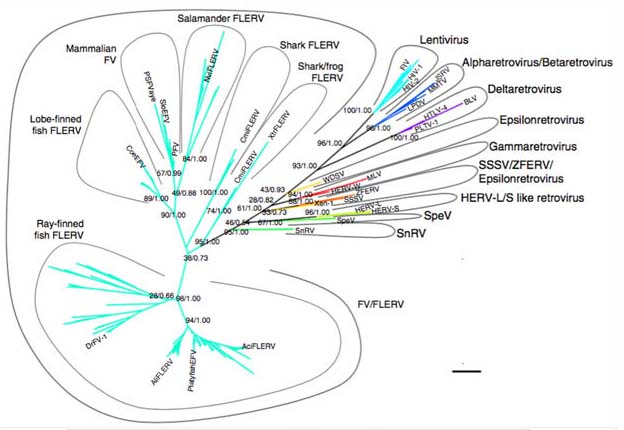

Fig 22b6b: Left: The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups (Koonin 2008). Picornaviruses are nonenveloped viruses that represent a large family of small, cytoplasmic, plus-strand RNA(~7.5kb) viruses, also responsibel for acture respiratory illness in humans. Compare fig 16b. Right: Retroviral phylogeny illustrating how foamy viruses (FVs) and amphibian and fish foamy-like endogenous retroviruses (FLERVs) relate to other retroviruses. These phylogenetic analyses suggest that this major retroviral lineage, and therefore retroviruses as a whole, have an ancient marine origin and originated together with, if not before, their jawed vertebrate hosts >450 million years ago in the Ordovician period, early Palaeozoic Era (Aiewsakun & Katzourakis 2017). An even older origin date for retroviruses can be inferred from the autonomous DNA transposon Polinton family parasitizing protists, funci and animals, which acquired a retroviral integrase at least 1 billion years ago ((Kapitonov & Jurka 2006 doi:10.1073pnas.0600833103).
HERV-K still has members with open reading frames and has been found to be expressed at the 8-cell stage of embryogenesis and appears to protect the embryo against infection from other viruses (Grow et al. 2015). HERV-K is transcribed during normal human embryogenesis, beginning with embryonic genome activation at the 8-cell stage, continuing through the emergence of epiblast cells in preimplantation blastocysts, and ceasing during human embryonic stem cell derivation from blastocyst outgrowths. Unlike most other human ERVs, HERVK retained multiple copies of intact open reading frames encoding retroviral proteins. It is transcriptionally silenced by the host, with the exception of in certain pathological contexts such as germ-cell tumours, melanoma or human immunodeficiency virus (HIV) infection. DNA hypomethylation at long terminal repeat elements representing the most recent genomic integrations, together with transactivation by OCT4 (which inhibits differentiation of stem cells in the pre-implantation embryo), synergistically facilitate HERVK expression in the early embryo from the 8-cell to blastula.HERV-K viral-like particles and Gag proteins can be found in human blastocysts, indicating that early human development proceeds in the presence of retroviral products. Expression of the HERV-K accessory protein Rec, may also inhibit viral infection. Moreover, Rec directly binds a subset of cellular RNAs and modulates their ribosome occupancy, indicating that complex interactions between retroviral proteins and host factors can fine-tune pathways of early human development.
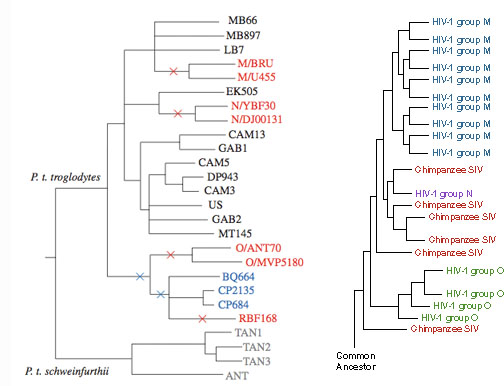 Fig 22c: The horizontally spread retrovirus HIV appears to have been transmitted to humans on three separate occasions leading to distinct evolving genotypes (Left: Sharp & Hahn 2010 - HIV genotypes red. Right: HIV groups M, N & O. evolution.berkeley.edu/evolibrary/article/medicine_04).
Fig 22c: The horizontally spread retrovirus HIV appears to have been transmitted to humans on three separate occasions leading to distinct evolving genotypes (Left: Sharp & Hahn 2010 - HIV genotypes red. Right: HIV groups M, N & O. evolution.berkeley.edu/evolibrary/article/medicine_04).
An X-linked complete version of HERV-K Xq21.33 probably capable of replication has been discovered, along with 36 undocumented provirus forms, in a search of the 1000 genomes project and a subset of the Human Genome Diversity Project panel (Wildschutte et al. 2016).
Mi and colleagues (2000) found a placental gene whose sequence was homologous to several retroviral envelope proteins. The sequence, now called syncytin, is identical to the envelope protein of the HERV-W retrovirus (Blond et. al.) which exists in around 40 apparently defective viral copies, including those in which the two syncytin viral env genes are fully functional (Mi et. al.). Syncytin is expressed at high levels in the syncytiotrophoblast (and at low levels in the testes) and nowhere else. Most of the other genes of the provirus have been mutated, suggesting that the envelope glycoprotein function was specifically selected. If cultured cells are made to express syncytin, they will fuse together, and this fusion can be blocked with antibodies against syncytin. HERV-W is only found in primates, but mice have similar retroviral blooms and ERV-related Syncytin genes have also been found in them (Dupressoir et. al.). The ability of mammals and thus ourselves to form a viable placenta and give birth to live young may thus depend on the mammals having harnessed a viral gene somewhere in our evolutionary lineage.
The fact that placental mammals depend on fusion between embryonic and maternal cells in the uterus and the fact that there are several different strains, e.g. in mice and men, which have clearly been 'borrowed' multiple times from retroviruses suggests an intimate evolutionary relationship between mammals and their retroviral counterparts. The discovery that syncytin is active in bone osteoclasts (Søe et al. 2011) and immature muscle cell myoblasts (Bjerregard et al. 2014), both of which involve cell fusion as well as the placenta, indicates a wider role. Redelsperger et al. (2016) discovered that knocking out both mouse versions, syncytin A and B, and is lethal, however knocking out only syncytin B results in scrawny males with depleted muscle fibres, showing that syncytin B is important in male muscle generation and appears to have a role in the greater muscle mass of many male mammals. Pivotally muscle is generated by the fusion of myoblasts to form multi-nucleated muscle fibres indicating a key role for syncitin nd why male mice have depleted muscle function, but why it is apparently confined to males remains to be resolved.
RNA expression of retroviral element RNLTR12-int (Retro-myelin) is crucial for myelination, by binding to SOX10 to regulate Mbp expression. It occurs in all jawed vertebrates through multiple convergent evolution events after initial speciation (Ghosh et al. 2024).
Retroviruses are divided between predominantly exogenous infectious habit such as HIV and SIV and conversion to endogenous transmission down the germ line such as the diverse HERV types. Magiorkinis G et. al. (2012) have verified that the loss of the Env gene which enables cell infection, is associated with super-amplification of germ-line retroviral elements by a factor of about 30, as exogenous retroviruses, switch to endogenous modes of selection. They have investigated the widespread occurence of retroviruses, including intracisternal A-particles, or IAPs, across the diversity mammal groups. Notably following Jern and Coffin (2008) the evolutionary tree of retroviruses both spans the vertebrates and includes both endogenous and exogenous habits, rooted in exogenous viral types.

Fig 23: (a) The seven retroviral genera: alpha-, beta-, gamma-, delta-, epsilon-, lenti-, and spuma-like retroviruses and their intermediate groups based on Pol sequences. Black branches indicate viruses known only in exogenous infectious forms (XRV); redbranches indicate viruses present in both XRV andendogenous (ERV) forms; and blue branches indicate ERVs Jern and Coffin (2008) . (b) Phylogeny of mammals with ERV megafamilies shown as colored circles (area is proportional to the percentage of the ERV loci in the genome represented by that family Magiorkinis G et. al. (2012). When retrotranspons are included (fig 22) they extend to all Eukaryote realms.
The defective copies of endogenous retroviruses may also serve to protect the host against further infection by becoming transcribed and causing incorporation of defective elements into the replicating virus (Best et. al.).

Fig 23b1: Left: Ediacarian fossils and evolution. Right: Cambrian Radiation with the Burgess Shale fossils.
The Ediacarian transition to the Cambrian Radiation, Homeotic Genes, Metamorphosis and Hybridization
One of the most stunning and puzzling aspects of evolution is the Cambrian radiation some 550 million years ago which over a very short period of geological time, gave rise to the major phylla of multicellular animals we see today. This radiation forms the core of the evolutionary tree of fig 1.

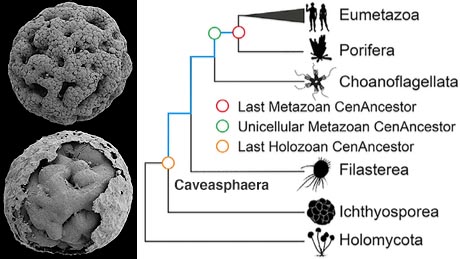
Fig 23b2 Left: (a) Dicksonia (b) Sprigginia (c) Yilingia (d) Charnia. Sprigginia and Yilingia (doi:10.1038/s41586-019-1522-7) show bilateral segmented structure
characteristic of early bilateran organisms leading to annelids, arthropods and vertebrates, while Dicksonia shows the quilted form of many enigmatic Ediacarian fossils.
Right: Caveasphaera develops within an envelope by cell division, ingression, detachment, and polar aggregation in a manner analogous
to gastrulation; together with evidence of functional cell adhesion and development within an envelope, this is suggestive of a holozoan affinity (Yin et al, doi:10.1016/j.cub.2019.10.057).Caveasphaera develops within an envelope by cell division, ingression, detachment, and polar aggregation in a manner analogous
to gastrulation; together with evidence of functional cell adhesion and development within an envelope, this is suggestive of a holozoan affinity (Yin et al, doi:10.1016/j.cub.2019.10.057).
The previous evolutionary epoch - the terminal Ediacaran (551–539 Mya) by contrast has far fewer and less elaborate fossil forms such as Charnia, Dicksonia and Spriggina, and particularly few organisms with well-preserved mineral skeletons. However the discovery of gastrula-ike embryonic states in Caveasphaera shows that embryonic features of single celled Ediacarian eucaryotes long pre-dated the emergence of multi-celled animals.
Variations of Ediacaran Morphology


Fig 23c: (left) Root of the animal tree with ctenophores being an ancient outlier and xenocoelomorphs forming a newly-discovered primitive phyllum (Rouse et al. 2016).
Right:
More recent evolutionary tree taking into account multiple genetic trends puts sponges back at the base division (Simion et al. 2017).
There have many proposals why such a rapid and abundant radiation could have occurred, including geological scenarios involving the ending of a snowball earth epoch in which the earth became frozen and thus reflected radiant heat, until rising CO2 levels caused a rapid thawing, setting off a major expansion of cyanobacteria, filling the atmosphere with oxygen and changing the ocean from an acidic state with dissolved iron and litle oxygen to one capable of harbouring diverse forms of multicellular life.
 Fig 24: Center to right: Homeotic genes specifying differentiation along the bodily axis have closely related sequences and are organized in a parallel scheme in arthropods and vertebrates and extend to cnidaria predating bilatera. Mutations such as antenapedia and bithorax in the fruit fly alter sequential specialization of segments. HOX genes share extensive sequence homology with phage lambda genes such as cro altering gene expression in prokaryotes (Luscombe et al. 2000 Genome Biology 1/1 reviews001.1–001.37). Upper left: The homeobox gene antennapedia which induces legs in the place of antennae in the fruit fly binding to DNA. Lower left: Ectopic compound eye on the leg of a fruit fly induced by mouse pax-6. Centre Left: Mutations of related genes in maize cause disruption of leaf development.
Fig 24: Center to right: Homeotic genes specifying differentiation along the bodily axis have closely related sequences and are organized in a parallel scheme in arthropods and vertebrates and extend to cnidaria predating bilatera. Mutations such as antenapedia and bithorax in the fruit fly alter sequential specialization of segments. HOX genes share extensive sequence homology with phage lambda genes such as cro altering gene expression in prokaryotes (Luscombe et al. 2000 Genome Biology 1/1 reviews001.1–001.37). Upper left: The homeobox gene antennapedia which induces legs in the place of antennae in the fruit fly binding to DNA. Lower left: Ectopic compound eye on the leg of a fruit fly induced by mouse pax-6. Centre Left: Mutations of related genes in maize cause disruption of leaf development.
However the underlying reasons may be genetic and more to do with the evolution of a pangenomic algorithm for generating the multicellular body plan based on homeotic and related developmental genes which are highly conserved and spread across the major animal and even plant kingdoms. Closely related schemes of homeotic genes drive the vertebrate and arthropod development along the bilateral axis, being involved in segmentation and notochord differentiation. For example pax-6, a gene involved in eye formation in the mouse, will induce ectopic eyes on the fruit fly, intriguingly the compound eyes insects usually have, indicating a deep commonality between the genes organizing the body plan in these two pivotal phylla.

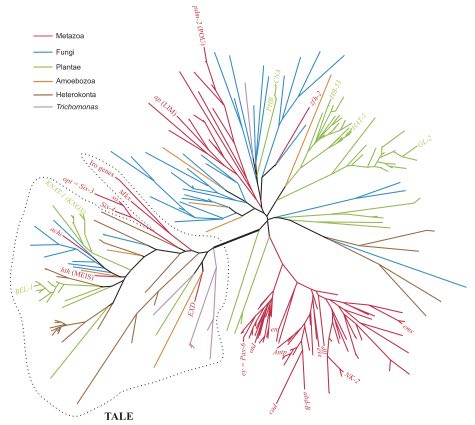
Fig 24b: Left: Increase in the ratio of noncoding DNA to total genomic DNA correlates with increasing biological complexity from 0.05 in Nanoarchaeum to 0.983 in Homo. Archaea in yellow, bacteria in blue, unicellular eukaryotes in black, the fungus Neurospora crassa in light grey, plants in green, non-chordate invertebrates in red, the urochordate Ciona intestinalis in yellow, and vertebrates in dark grey. It is key to understanding how higher organisms, through to ourselves, were able to evolve, because single base pair mutations of non-coding DNA are less disruptive, as they don't have to be translated and make only a differential change to RNA and promoter binding. Complementing this, natural and particularly sexual selection is both phenotypic and conscious (Taft & Mattick 2003). This means that humans have only 2.82 times the coding genome of the amoeboid slime mould Distyostellium discoideum and 2.7 times as many genes, with only 2.0 and 1.57 as many compared with the simple roundworm Caenorhabditis elegans. Evolution has thus become a symphony on largely the same genes, as is also made clear in the great archaean diversification 3 bya ago. Centre: Presence and abundance of transcription factors (TFs) in eukaryotes, including homeotic genes are consisent with an origin spanning both metazoa and sincle ceelled eucaryotes going back to the LECA root. The heat map depicts absolute TF counts according to the color scale. TFs/DBDs (rows) are clustered according to abundance and distribution, and species (columns) are grouped according to phylogenetic affinity. Major eukaryotic lineages are indicated (de Mendoza et al. 2013). Right: Maximum Likelihood phylogenetic analysis of eukaryote homeodomains (HDs) spans single celled and multicelled eucaryotes (Derelle, R. et al. 2007).
This suggests it may have taken evolution a considerable time to come up with such an algorithmic regulatory process, but that once it came into play, it permitted almost symphonic variations, leading to the diversity of the major animal phylla over a relatively short geological time.
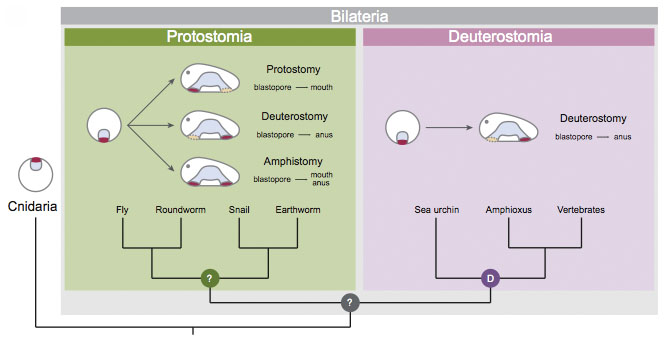
Fig 25: (Left) Body schemes of the bilateria (Martın-Duran et al. 2012). (Right). Anciently conserved synteny across bilaterians, sponge, and cnidarians. (Right) Numbered horizontal bars represent the chromosomes of five species. (Left) Phylogenetic tree is shown with the root marked by a black circle. Sponge is shown in a central location to display conserved syntenies with both bilaterians (top) and cnidarians (bottom). Common names and three-letter acronyms for each species are shown (see text for details). On the right, colored vertical lines connect orthologous genes across the five species. Only connections between chromosome pairs with significantly enriched conservation of synteny are shown. Each color represents a distinct ALG (ancestral linkage group). Two or more colors converging on a chromosome (e.g., amphioxus BFL5 and hydra HVU6) indicate fusion-with-mixing of ancestral units (Simakov et al. 2022) .
A central division in the Cambrian radiation is the one that led to the bilateria - organisms with the left right symmetry possessed by both arthropods and vertebrates. The conventional argument is that the founding event differentiating bilateria from earlier organisms such as the cnidaria which have a mouth only is the symmetry introduced by the formation of an anus and an intestinal tube. Here things get complicated because embryonic development can go either from mouth first, based on the blastopore originating in cnidaria and then to anus or vice versa. In the conventional division the deuterostomes including the vertebrates go "arse-first" i.e. anus > mouth but the proterostomes go mouth > anus. The difficulty is that key deuterostomes such as the priapulid worm Priapulus caudatus, which was abundant in the middle Cambrian, actually develops on a deuterstome plan although in terms of molecular evolution the expression of bra, cdx, foxA, gsc, and otx during early development is similar to nematodes and arthropods spanning the "skin-shedding" ecdysozoa. Given the fact that the Chaetognatha or arrow worms also have this pattern the deuterstome pattern appears to be ancestral with the proterostomes forming a diverse set of body plans (Martın-Duran et al. 2012).

Fig 25b: (Left) Hypothetical origin of nerve cords from a common ancestor requires an inversion fo the arrangement from ventral to dorsal and now appears to be inconsistent with a more complete picture of the diversity of transitional organisms, implying a variety of origins of central nerve cords (right) through convergent evolution utilizing the same suites of underlying developmental genes (Martin-Duran et al. 2017).
The unique origin of a bilaterian nerve cord from a common ancestor was originally conceived in 1875 by Anton Dohrn noting anatomical similarities between the central nerve cord of annelids and vertebrates as resulting from a common ancestor of the proterostomes and deuterostomes, which flipped from a ventral to a dorsal position in the latter group, leading to vertebrates. This idea appeared to be confirmed when the same genes involved in the development of vertebrate central nerve cords were found to also be activated in the nerve cord of the fly Drosophila melanogaster and the marine annelid Platynereis dumerilii. However this position has been challenged as a result of research, in which firstly it was found that a critical developmental protein bmp is activated in hemichordate acorn worms early in their development - well before they form the two nerve cords that run along the sides of their bodies, (Lowe et al. 2006), suggesting that the common ancestor of hemichordates and chordates did not use its Bmp-Chordin axis to segregate epidermal and neural ectoderm but to pattern many other dorsoventral aspects of the germ layers, including neural cell fates within a diffuse nervous system. More recently, a team led by Andreas Hejnol, (Martin-Duran et al. 2017) surveyed a variety of organisms ancestral to both groups, including Xenacoelomorpha (Maxmen 2011), rotifers and others and have found that cordal nervous systems have arisen multiple times from a variety of transition states, from the nerve nets of cnidaria to the central nervous systems and focal ganglia of vertebrates and arthropods.
The conventional theory of insect morphogenesis is the evolution from eggs giving rise to small adult form individuals to delayed maturation of embryonic forms in the form of larvae, which did not compete with the adults in food consumption and habit, thus leading to a two-stage life cycle, with non-competing foraging larval and reproductive adults forms (Jabr). A controversial theory (Ryan) posits that aspects of metamorphosis, which we also see in insects, but  more pivotally in marine organisms such as echinoderms, may have resulted from early hybridization even between organisms which have now become distinct phylla, such as vertebrates and echinoderms. For example Nectocaris pteryx appears to have a body plan looking like a chimera of an arthropod head and the abdomen of an entirely different phyllum, although this may be a result of the way fossils are depicted in drawings and this species may simply be an early cephalopod (Smith & Caron). The validity of this idea, at least in insects, is highly controversial and hotly disputed (Hart & Grosberg, Williamson).
more pivotally in marine organisms such as echinoderms, may have resulted from early hybridization even between organisms which have now become distinct phylla, such as vertebrates and echinoderms. For example Nectocaris pteryx appears to have a body plan looking like a chimera of an arthropod head and the abdomen of an entirely different phyllum, although this may be a result of the way fossils are depicted in drawings and this species may simply be an early cephalopod (Smith & Caron). The validity of this idea, at least in insects, is highly controversial and hotly disputed (Hart & Grosberg, Williamson).
Fig 26: Fossil and two very different scientific illustrations (insets) of Nectocaris ptaryx
and two views of the larva of Luidia sarsi emitting an echinodermal 'offspring'.
Studies attempting to trace the evolutionary tree depending on the simpler larval forms from which one would expect the adult form to have evolved have yielded contradictory results, suggesting the two might have independent genetic origins. Genetic analysis of sea squirts, which have vertebrate larval forms with a notochord and a primitive brain but metamorphose into fixed sea-floor feeders, suggests they have two genomic components, one coming from vertebrates and the other from and an unknown but now extinct non-vertebrate at a very early stage in the evolution of animals. Echinoderms themselves have larvae with a bilateral body plan which later becomes colonized by pluripotent cells in the abdominal cavity forming radially symmetric organisms which grow into adults. In the starfish Luidia sarsi, the embryonic form some 4 cm long survives for several months as a vegetarian living off phytoplankton after its starfish 'offspring' have burst out to their carnivorous habit of hunting other starfish.
Donald Williamson, who in the 1950s advanced the 'larval transfer' theory claimed to have successfully hybridized fertilised eggs from the sea squirt Ascidia mentula with sperm from the sea urchin Echinus esculentus. Then in 2002, in an unpublished study with Sebastian Holmes and Nic Boerboom, he did the reverse cross, using eggs from the urchin and sperm from the sea squirt. Both crosses resulted in large numbers of offspring, the majority of eggs developing into easel-shaped larvae - the 'pluteus' form typical of sea urchins, rather than the tadpole larvae that are the hallmark of sea squirts. Most of these larvae subsequently metamorphosed to a rounded adult form, which Williamson called a 'spheroid'. The first cross created spheroids with a suction cup, that enabled them to attach to surfaces. Most intriguingly, the second produced spheroids that reproduced asexually through budding, the pinching off of a section of the body to create a clone. However these were never subjected to genetic analysis. A cross between two echinoderm species has however resulted in new developmental phenotypes with confirmed hybridized genomes.
Evolution of Plants and Chloroplasts
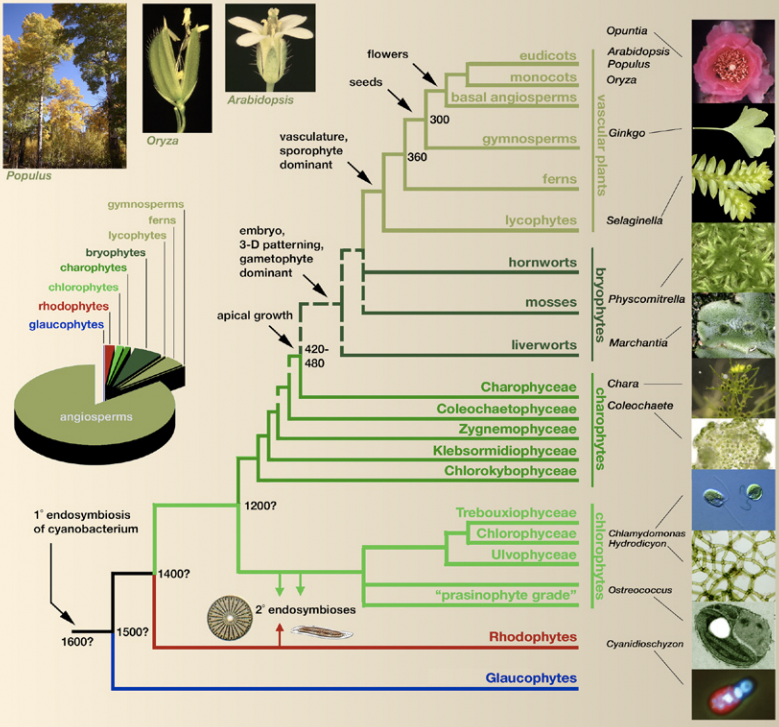

Fig 26a: Trees of the root of Plantae (Bowman et al. 2007) and of the Seed-bearing plants (Lee at al.. 2011).
The emergence of the land plants (Cheng et al. 2019) has been found to pass through single-celled and simple fibre-forming colonial Spirogloea muscicola, a type of Zygnematophyceae and Mesotaenium endlicherianum, which picked up bacterial genes, rather than Carophytes, which look like underwater plants. These horizontal transfers involved genes which help plants to survive droughts and other kinds of stress. Even today, land plants rely on the genes to make spores and seeds that can survive for months or years in dormancy. The researchers found no similar versions of these genes in other algae. But they are present in bacteria that live in soil. The algae make a spongy coat to soak up water, and some bacteria feed on the carbohydrates that make up the coat. In return, they produce vitamins that the algae may need. This intimate connection may have allowed genes from the bacteria to slip into the algae's DNA.

Fig 26a1: Emergence of Land plants (Cheng et al. 2019).
Three studies have elucidated the evolutionary relationships of the major plant phylla leading up to the seed-bearing and flowering plants. Above are shown the first two, which despite a wave of genetic sequencing still have some ambiguities, as noted in the left figure. Key is the radiation of al land plants from a relative of the green algae charophyta exemplified by the freshwater alga chara which displays sexually differentiated reproductive organs (inset in right figure)producing sperm and ova as in all plants up to and including the gymnosperms.


Fig 26a2: Left: Evolution of the flowering plants showing the structure of the bisexual ancestor flower (Sauquet et al. 2017). Right: Evolution of the chloroplast (Keeling 2004, Raven & Allen 2003, Rockwell et al. 2014). Plastids have been directly incorporated from Cyanobacteria only twice once a billion years ago and again 60 million years ago a Synechococcus-like cyanobacterium was incorporated into Paulinella chromatophora with the subsequent loss of its pseudopods (Mackiewicz et al. 2012). Red algae subsequently became incorporated into a variety of chromalveolate protists , with green algae being incorporated into Euglena. The euglenoid trypanosomes still show evidence of previous photosynthesis (Hannaert et al. 2003).
According to Sauquet et al. (2017), the ancestral flower was bisexual, with both female (carpels) and male (stamens) parts, and with multiple whorls (concentric cycles) of petal-like organs, in sets of threes. About 20% of extant flowers have such 'trimerous' whorls, but typically fewer: lilies have two, magnolias have three. Ancestral state reconstruction of functional sex of flowers in angiosperms: the study results show that bisexual flowers are ancestral and that unisexual flowers evolved many times independently; the pie charts at the center of the figure indicate the proportional likelihoods for reconstructed ancestral states at 15 key nodes. The team also reconstructed what flowers looked like at all the key divergences in the flowering plant evolutionary tree, including the early evolution of the two largest groups of flowering plants: monocots (e.g., orchids, lilies, and grasses) and eudicots (e.g., poppies, roses, and sunflowers).


Fig 26a3: Two views of fungal evolution qualitative and genetic investigating gene fission and fusion (Leonard & Richards 2012).
Evolutionary tree of Psilocybes and related groups <––> Psilocybes of Mexico
Fungal evolution has seen diversification into a diverse spectrum of species. Although only about 100,000 have so far been identified, compared with 310,000 plant species, the true number is estimated to be 1.5 million, second only to the 5 million estimated species of insects. Above are shown two evolutionary trees the first giving key qualitative milestones in the fungal radiation and the second a more detailed genetic analysis focusing on gene fissions and fusions. Both trees emphasize the fact that Chitrids, which produce motile gametes lie at the root of the fungal tree (Leonard & Richards 2012). Lutzoni et al. (2004) have produced an even more detailed tree.
Evolution of Arthropods and Insects



Fig 26a4: Evolutionary trees of the arthropods (Regier et al. 2010), insects (Misof et al. 2014) and butterflies (Espeland et al. 2018).
Evolutionary tree of all 845 species of US and Canadian butterflies (Zhang et al. 2019 bioRxiv doi:10.1101/829887).
The genomes of butterflies and moths have remained largely unchanged for more than 250m years despite their enormous species diversity. Lepidoptera are among the most diverse animal groups known to science, making up approximately 10% of living organisms on Earth.

Fig 26a5: Phylogenetic relationships of 210 lepidopteran species and the distribution of large-scale rearrangement events (Wright et al. 2024).
Insects arose from fresh-water hexapod crustaceans similar to water fleas and fairy shrimp around 410 million years ago shortly after the colonization of the land by vascular plants around 450 million years ago (Glenner et al. 2006). Early are coneheads and springtails and the May Fly, whose nymph stage is still aquatic and still has seven rows of gills and a posture similar to a crustacean (Regier et al. 2010, Misof et al. 2014).

Fig 26a6: Evolution of Birds (Stiller et al. 2024, Mirarab et al. 2024)
The authors observed sharp increases in effective population size, substitution rates and relative brain size in early birds, shedding new light on the adaptive mechanisms that drove avian diversification in the aftermath of the cataclysmic mass extinction event that wiped out the dinosaurs 66 million years ago. With the aid of their advanced computational methods, the researchers were also able to shed light on something unusual that they had discovered in one of their previous studies: a particular section of one chromosome in the bird genome had remained unchanged for millions of years, void of the expected patterns of genetic recombination.
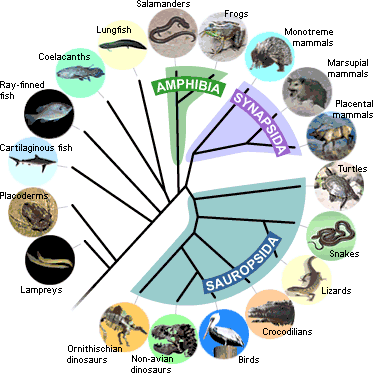


Fig 26b: Left: Simplified tree of Vertebrate Evolution by major classes. Centre: Penetrative Sex (John Long, David Choo). Right: Evolution of cichlid fish (Ronco et al. 2020).
Vertebrate Evolution, Parental Care and Penetrative Sex
Above left is shown a simplified evolutionary tree of the vertebrates, showing the major classes and noting the point of divergence between the symapsids leading to mammals and the sauropsids leading to birds.
Dark DNA: Vertebrate Evolution at a Crossroads?
Recent fossil evidence suggests that penetrative sex has evolved four times in the vertebrate tree and was the original form of sex in the ancient placoderms that gave rise to all the jawed vertebrates (Long et al. 2014). It still appears in stingrays, although they have lost the bony claspers and replaced tham with cartilagenous ones, and has re-emerged in both guppys and land vertebrates which have evolved a variety of intromittent penises. This suggests that fundamental components of development have been preserved for long epochs leading to penetrative sex not having a developmental gene bootstrap.

Fig 26b1: Evolution of parental care. Left: Parental care in a 520 million-year-old stem-group euarthropod (Fu et al. 2018). Centre: Mapping parental care on a general phylogeny of the major amniotic groups shows that post-natal parental care appears to be the ancestral condition for all amniotes. Right: Post-natal parental care in a Cretaceous 125 million-year-old diapsid (Lu et al. 2015).
Parental care has also been shown to have an early origin. Although fossils demonstrating this are rare, fig 26b1 shows two instances, one of a Cambrian arthropod surrounded by offspring and the other of a Cretacious diapsid. Post-natal parental care has been calculated to have been the acestral condition of amniotes, the tetrapod vertebrates comprising the reptiles, birds, and mammals.
Evolution of bony (Betancur-R et al. 2017) and ray-finned (Hughes et al. 2018) fishes .
 Fig 26b2: Conflict in the tree of mammalian diversification. Detailed traditional DNA-based evolutionary tree of the mammals (right) (Meredith et al, dos Reis et al) tends to have a different order of diversification from one based on the number of new miRNAs appearing in successive branches (top left Dolgin). Micro miRNA numbers have also been suggested to be correlated with neural complexity (bottom left Technau). Larger image of the right figure. Major mammal groupings image.
Fig 26b2: Conflict in the tree of mammalian diversification. Detailed traditional DNA-based evolutionary tree of the mammals (right) (Meredith et al, dos Reis et al) tends to have a different order of diversification from one based on the number of new miRNAs appearing in successive branches (top left Dolgin). Micro miRNA numbers have also been suggested to be correlated with neural complexity (bottom left Technau). Larger image of the right figure. Major mammal groupings image.
Mammalian Radiative Adaption: Traditional DNA versus Micro RNAs
Different assay methods are shedding an intriguing light on radiative adaptation and diversification of animal species, from the Cambrian through to the present day. The detailed branching of the tree of life when calculated by traditional mutational DNA methods (Meredith et al, dos Reis et al), appears to differ significantly from a new technique developed by Kevin Peterson (Dolgin) that depends on the number of newly accrued micro miRNAs which modulate gene expression by selectively binding to specific messengers inhibiting their expression.
A single miRNA is thus able to modulate the expression of a diverse array of mRNAs to which it binds, thus providing for sophisticated forms of coordinated regulation conducive to phylogenetic complexity. It has also been suggested that neuronal complexity correlates with the number of miRNAs (Technau, Grimson et al) an interesting question in itself to do with how complex nervous systems are generated in development. Notice here that humans have fewer protein genes than a mouse, roughly 21,000 against 22,000 although we have a brain with 10,000 times as many neurons, so we need to have an idea how organismic complexity evolves in terms of sophisticated gene regulation, and miRNAs do just that.
Consitent with such a role in multi-celled evolution, the appearance of miRNAs goes back to the earliest multicelled animals. Sea anemones already carry up to 40. Metazoa, from sponges to bilateria, also share the two classes of piRNA, the second of which plays a role in suppressing transposable elements in gametogenesis, by containing a sequence complementary to a transposase mRNA. In fruit flies these are directed against DNA transposons, but in mammals they target LINE L1 and IAP transcription during meiosis in the germ line, by methylating L1 and IAP DNA sequences (Aravin et al).

Left: Lineage-specific primate evolutionary tree Bi et al., (2023) Sci. Adv. 9, eadc9507 Right: Tree of primate masturbation Brindle et al. (2023) doi:10.1098/rspb.2023.0061
While a traditional DNA-based tree places primates and humans much closer to rodents, as highly evolved branches, with the elephants diverging earliest, an miRNA analysis places rodents as branching out earliest, something which might seem to be consistent with their possibly closer correspondence to the founding shrew-like mammalian type. The critical question determining the fate of the miRNA perspective is what the rate of loss of these small RNA molecules is in evolution. A higher rate of loss would tend to remove the inconsistency. While the picture is consistent with retaining miRNAs in mammalian diversification, in insects and a primitive chordate sudden losses have occurred.
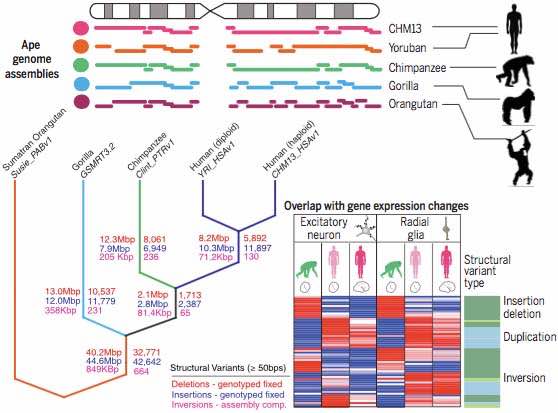 High resolution evolutionary tree of great apes (Kronenberg et al doi:10.1126/science.aar6343).
High resolution evolutionary tree of great apes (Kronenberg et al doi:10.1126/science.aar6343).
Kronenberg et al 2018 (doi:10.1126/science.aar6343) describe new great-ape genome assemblies, generated using a technology that surpasses previous methods. This work marks a new stage in our ability to study and compare these species. . Structural variation in the genome is important, particularly on the short evolutionary timescale that separates humans and other great apes, because it provides a way for genomes to evolve rapidly. When a whole chunk of DNA is removed or duplicated, its molecular function can be inhibited or enhanced in one step, rather than through successive mutations at individual bases. Much of the great-ape genome seems to be modular in nature, and is therefore susceptible to these kinds of changes. It has also been discovered that gene loss is a key mechanism for evolutionary change (Sharma et al. 2018 Nature Commun. doi:10.1038/s41467-018-03667-1). This might seem counterintuitive, but genes often act to constrain, rather than promote, a particular function. Disabling them by removing, duplicating or relocating a chunk of DNA might be the simplest way to confer beneficial effects. The authors found about 600,000 structural differences between these genomes and that of humans, including more than 17,000 differences specific to humans.
Of these, many changes disrupt genes in humans that are not disrupted in other apes. Genes whose activity is suppressed specifically in humans are more likely than other genes to be associated with a human-specific structural variant. Many genes produce multiple versions, called isoforms, of the protein they encode, each of which can have a different role. The researchers found evidence that a large deletion in the gene FADS2 involved in the synthesis of fatty acids needed for brain development and immune response might have altered the distribution of isoforms the gene produces. These are difficult to obtain from a purely herbivorous diet and FADS2 has been a target for natural selection associated with dietary changes towards or away from animal fats in recent human evolution. Structural variation also seems to have had a role in brain evolution. Human brains are much larger than those of other apes, and it is plausible that genes involved in brain growth and development were key to the evolution of this trait. These analyses revealed that 41% of genes whose activity is suppressed in human radial glial cells are associated with a human-specific structural variant.
For modern lineages of birds and mammals, few fossils have been found that predate the Cretaceous–Palaeogene (K-Pg) boundary, although molecular studies using fossil calibrations have shown that many of these lineages existed at that time. One intriguing way of checking the evolution of mammals and its timing is to examine the parasites of birds reptiles and mammals and to develop a "tree of lice". Smith et al. (2011) demonstrate that the major louse suborders began to radiate before the K-Pg boundary, lending support to an earlier Cretaceous diversification of many modern bird and mammal lineages.


Fig 26c: Left: Evolutionary tree of mammalian male infanticide (circled species) which occurs in around half of the 200 species invesigated. It is commonest in social species (dark grey) and less so in solitary species (light grey) and even less in monogamous species (black) (Lukas & Huchard 2014) Right: Since it does not contribute directly to reproduction, same-sex sexual behaviour is considered an evolutionary conundrum. According to currently available data, this behaviour is not randomly distributed across mammal lineages, but tends to be particularly prevalent in some clades, especially primates. Ancestral reconstruction suggests that same-sex sexual behaviour may have evolved multiple times, with its appearance being a recent phenomenon in most mammalian lineages. Our phylogenetically informed analyses testing for associations between same-sex sexual behaviour and other species characteristics suggest that it may play an adaptive role in maintaining social relationships and mitigating conflict (Gómez et al. 2023).
Mammals give birth to live young and lactate. This has caused the reproductive investment of the two sexes to become highly skewed, with males investing primarily in fertilization, while females are investing primarily in parenting. This has a variety of consequences. For example only around 3% of mammal species are socially monogamous, with the rest being either polygynous with harems, or having promiscuous females. This again skews the reproductive strategies further, because there are secondary consequences. Females are less likely to go in heat and become pregnant while they are lactating a brood, and offsping of other males will compete with his own, so there is a double investment in species with competing males, to kill the offspring of competitors - male infanticide. Around half of mammalian species, including the ancestors of the great apes and humans, are inveterate infanticiders, as promiscuous chimps and harem forming gorillas are, and the trait appears to have evolved multiple times. In turn, the females adapt to promiscuous mating, often with an advertised estrus to make it as difficult as possible for males to determine paternity (Lukas & Huchard 2014). Finally the males adapt by forming larger testes to deal with the issues of sperm competition involved in promiscuity.
Emergence and Diversification of Modern Humans
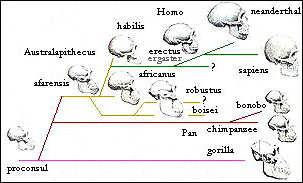 Fig 27: One hypothetical evolutionary tree for humans and related apes. There is much debate about the actual form of such a tree.
Fig 27: One hypothetical evolutionary tree for humans and related apes. There is much debate about the actual form of such a tree.
Homo sapiens appears to have evolved into a single dominant species on the planet, after preceding period in which fossil evidence suggests there were several different anthropoid species coexistent.
The final stage of this process was the disappearance of Homo erectus and Neanderthal, the latter after a well-defined period of coexistence in Europe at the end of the last ice age.
Our own evolutionary and cultural roots appear to lie in Africa, with evidence of culture and cosmetics running back over 100,000 years, in addition to evidence for tools and weapons. An alternative regional development theory has proposed that humans evolved through a considerable amount of interbreeding over the whole African and Asian continental region, however genetic evidence is coming to point towards an African origin with only at most very occasional cross fertilization with related species.
 Fig 28: Human-Neanderthal-Chimp divergences (Green et. al. 2006)
Fig 28: Human-Neanderthal-Chimp divergences (Green et. al. 2006)
According to genetic analysis, Neanderthals diverged from homo sapiens ~500,000 years ago. There has been no major interbreeding, but possibly some transfer of genes e.g. from human males to Neanaderthal females, although candidate human genes conferring natural advantage do have a profile consistent with transfer from Neanderthals (Pennisi).
Specific genes such a s PDHA1 consist of two families with the last common ancestor 1.8 million years ago, and microcephalin variants appearing 40,000 years ago also have differences suggesting an original divergence 1 million years ago suggesting 'introgression' from Neanderthals (Jones). An even more ancient divergence in the pseudogene RRM2P4 in East Asian people suggests interbreeding with Homo Erectus. Some evidence from skeletons is also consistent with this picture. However more recent sequencing of the Neanderthal nuclear genome (Callaway 2008) suggests little or no interbreeding with Homo sapiens and has cast doubt on the existence of the microcephalin variant in Neanderthals, as well as a gene associated with increased fertility in Icelanders also attributed to transfer from Neanderthals.

Fig 29: The "Out of Africa" hypothesis may be consistent with a degree of regional development
involving some sexual interbreeding with Neanderthals and Homo erectus (New Scientist).
Comprehensive investigation of the Neanderthal genome (Green et al. 2010, Dalton 2010) suggests that there was a period of interbreeding between Neanderthals and humans in the Near East around the time of the first migration out of Africa, rather than more recently in Europe, as the putative sequences are shared by non-African French, Han and Papuan, but not by the African Yoruba or San. It is estimated that among the former, 1-4% of the genome derives from Neanderthal sequences, although there is little evidence for these corresponding to the specific genes suggested by Lahn's team. Other transfers could have occurred but are not apparent in the research.
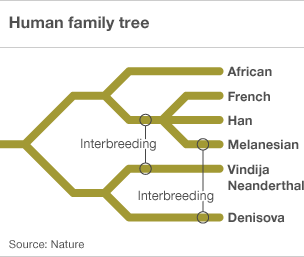 The situation has been complicated by two finds. Firstly we have the 'hobbit' human remains found on Flores, named Homo fiorensis. These are variously claimed to be a Separate human species possibly related to Homo erectus, or disclaimed as microcephalic human pygmy peoples. More recently we have the discovery of remains of Denisovans (Callaway 2010), a further species branching off from the Neanderthals and their lines branching from Homo sapiens some 800,000 years ago. Genetic analysis of the remains indicates a significant interbreeding specifically with Melanesian people of some 6% (Reich et. al 2010, Meyer et al 2012).
The situation has been complicated by two finds. Firstly we have the 'hobbit' human remains found on Flores, named Homo fiorensis. These are variously claimed to be a Separate human species possibly related to Homo erectus, or disclaimed as microcephalic human pygmy peoples. More recently we have the discovery of remains of Denisovans (Callaway 2010), a further species branching off from the Neanderthals and their lines branching from Homo sapiens some 800,000 years ago. Genetic analysis of the remains indicates a significant interbreeding specifically with Melanesian people of some 6% (Reich et. al 2010, Meyer et al 2012).
More recent investigations show that interbreeding with other hominins was critical to the globalization of Homo sapiens. Human leukocyte antigens (HLAs), a family of about 200 genes that essential to our immune system also contains some of the most variable human genes: hundreds of versions - or alleles - exist of each gene in the population, allowing our bodies to react to a huge number of disease-causing agents and adapt to new ones. One allele, HLA-C*0702, is common in modern Europeans and Asians but never seen in Africans; Peter Parham has found it in the Neanderthal genome, suggesting it made its way into H. sapiens of non-African descent through interbreeding. HLA-A*11 had a similar story: it is mostly found in Asians and never in Africans, and Parham found it in the Denisovan genome, again suggesting its source was interbreeding outside of Africa. This tallies with interbreeding giving H. sapiens pivotal resistance to non-African diseases. While only 6 per cent of the non-African modern human genome comes from other hominins, the share of HLAs acquired during interbreeding is much higher. Half of European HLA-A alleles come from other hominins, says Parham, and that figure rises to 72 per cent for people in China, and over 90 per cent for those in Papua New Guinea (Marshall 2011).
The contribution to immunity from Neanderthals has now become strongly evident, with highly significant differences between immune response to pathogens in both macrophages and monocytes in separate studies highlighting differences between African and non-African populations, which involve genes with Neanderthal homology, implying a Neanderthal-derived shift in the immune response to cope with new kinds of pathgens in new areas the migrants found themselves in. The greater intensity of immune response in African populations may also may explain the three-fold higher rates of auto-immune disease in African women (Reardon 2016).
In a 2014 study by David Reich and coworkers, genes for keratin filaments that lend toughness to skin, hair and nails, were enriched with Neanderthal DNA. This may have helped provide the newcomers with thicker insulation against cold conditions, the scientists suggest. But other genes are implicated in human illnesses, such as type 2 diabetes, long-term depression, lupus, billiary cirrhosis - an autoimmune disease of the liver - and Crohn's disease. Other regions of the human genome, including the X-chromosome, are devoid of Neanderthal sequences suggesting they were selected against as deleterious. A genome region that lacked Neanderthal genes includes FOXP2, thought to play an important role in human speech (Sankararaman et al 2014 , Vernot & Akey 2014).
More recently selection for specific Tol-like receptors significant in disease resistance and acclimatization to high altitudes in Tibetans have both been tied to Neanderthal allelles among a wide survey of relative contributions to disease related genes (Callaway E 2015). In a 2016 study traits linked to hypercoagulation, depresion and tobacco addiction correlated with Neanderthal alleles. Although these may be disadvantageous in older modern populations they must have either conferred advantages during reproductive age or general advantages for the population of the time. Coagulation may have protected against injury but makes people more prone to stroke in modern populations (Simonti et al. 2016). A second 2016 study by Akey and co-workers confirms that hybridization with Neanderthals and Denisovans provided an important reservoir of advantageous mutations for modern humans that enabled adaptation to emergent selective pressures as they dispersed out of Africa. Our results show that immune and pigmentation traits were frequent substrates of adaptive introgression and that in many cases adaptive archaic haplotypes also contribute to the disease susceptibility in contemporary individuals. Many positively selected archaic haplotypes act as expression quantitative trait loci, which modulate the quantitative expression of particular genes, suggesting that modulation of transcript abundance was a common mechanism facilitating adaptive introgression (Gittelman et al. 2016).
Three studies examining immune response to infection highlight such differences. Nédélec, Y. et al. (2016) measured how gene expression in macrophages changed in response to the infection. About 30% of the approximately 12,000 genes that they tested were expressed differently between the two groups, even before infection and many of the genes whose activity changed the most during the immune reaction had sequences that were very similar between Europeans and Neanderthals, but not Africans. Quach, H. et al. (2016) grew monocytes in a dish and infected them with bacteria and viruses. Once again, the two groups showed differences in the activity of numerous genes, and Neanderthal-like gene variants in the European group played a major role in altering their immune response. The differences were especially stark in the way that those of African descent and the other half of European descent responded to viral infection. For some diseases, such as tuberculosis, a lower immune response tends to help with survival, and modern humans in Europe adopted the Neanderthal traits that helped with this. Overactive immune systems could help to explain why African American women, for instance, are up to three times more prone to the autoimmune disease lupus than white Americans. Long, frequent - and more likely adaptive — segments of Neanderthal ancestry in modern humans have been found to be enriched for proteins that interact with viruses (VIPs). VIPs that interact specifically with RNA viruses were more likely to belong to introgressed segments in modern Europeans. Also showing that retained segments of Neanderthal ancestry can be used to detect ancient epidemics (Enard & Petrov 2018). The researchers previously found that VIPs evolve under both stronger purifying selection and tend to adapt at much higher rates compared to similar proteins that do not interact with viruses. They estimated that interactions with viruses accounted for $30% of protein adaptation in the human lineage (Enard et al. 2016). Resistance to eucaryote parasites such as malaria was also found (Ebel et al. 2016).
The oligoadenylate synthetase (OAS) locus, which consists of three genes - OAS1, OAS2, and OAS3 - that encode enzymes involved in the innate immune response against viruses, and are among the core genes that are important to stop viral replication, A further comparison of OAS sequences among human populations revealed that this OAS Neanderthal allele is found in about 60 percent of individuals in Africa. However, outside of Africa, it is only found in individuals that harbor the Neanderthal haplotype. It is likely that Neanderthals were better adapted to the pathogens present in non-African environments than anatomically modern humans that had newly moved into these regions and it appears that this allele was lost during the out-of-Africa migration and that the Neanderthal haplotype resurrected this allele after the bottleneck following the human migration out of Africa (Sams A et al. 2016). The Vindaja specimen has added some 10% more Neanderthal genes to the introgressed complement including genes for LDL cholesterol levels, and schizophrenia (Prüfer et al 2017).
McCoy, Wakefield and Akey (2017) note that there is significant downregulation of Neanderthal genes in both the brain and testes, indicating that these genes are mildly deleterious: "Recent theoretical work predicts that Neanderthals suffered a high load of weakly deleterious mutations accumulated during extended population bottlenecks. Assuming additive fitness effects, this mutational burden was estimated to have reduced Neanderthal fitness by at least 40% compared to modern humans. Under this model, deleterious haplotypes introgressed into larger modern human populations would have been subject to strong selection during the first ~20 generations after hybridization — a prediction with growing empirical support from genetic data. Nevertheless, many weakly deleterious variants are predicted to persist in present-day human populations, with a cumulative impact comparable to that of the Out-of-Africa bottleneck. Contributing to this result, we observed a striking bias toward downregulation of Neanderthal alleles in the brain and testes. Brain regions had significantly lower expression of Neanderthal alleles than non-brain tissues, particularly in the neuron-rich cerebellum and basal ganglia regions. This level of downregulation is exceptional, as equalsized samples of non-introgressed SNPs matched for sample sizes of individuals and tissues showed no such bias. Further consistent with these data, brain regions including the cerebellum were enriched for significantly down-regulated compared to significantly upregulated Neanderthal SNPs. Significant downregulation of introgressed alleles in the brain is particularly remarkable given the previous observation that brain-expressed genes show less alelle-specific expression (ASE) overall, a finding that was attributed to reduced levels of genetic diversity in this gene set. One brain-specific gene that exemplifies this pattern of down- regulation is NTRK2, which encodes a neurotrophic tyrosine receptor kinase that regulates neuron survival and differentiation as well as synapse formation."
Neanderthal versions of genes in the testes, including some needed for sperm function, were also less active than human varieties. That finding is consistent with earlier studies that suggested male human-Neandertal hybrids may have been infertile. But Neandertal genes don't always lose. In particular, the Neandertal version of an immunity gene called TLR1 is more active than the human version. Lopsided gene activity may help explain why carrying Neandertal versions of some genes has been linked to human diseases, such as lupus and depression.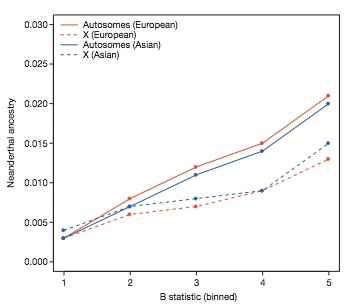
Functionally important regions are deficient in Neanderthal ancestry (Sankararaman et al 2014).
A study of the Neanderthal Y chromosome, which is probably extinct in the human population, despite interbreediing, has shown that several key male histocompatability genes on the Y are mutated in a way which could have led to a maternal immune response and miscarriages, forming a barrier to interfertility (Mendez F et al. 2016). Moreover there are signs of hybrid infertility, suggesting only about 1 in 50 inter-matings resulted in fertile offspring, as regions in the X-chromosome, and on other chromosomes linked to testis genes, and mitochondrial DNA are all devoid of Neanderthal genes, a pattern common to interspecies infertility. Evidence from a study of European Ice Age genomes shows the proportion of Neanderthal DNA has been declining due to natural selection (Fu et al. 2016).
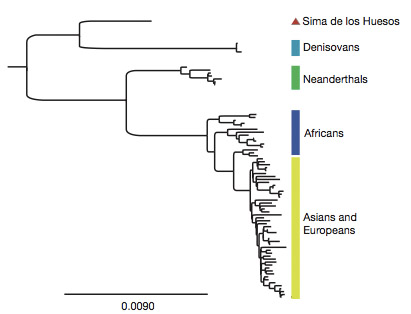 Fig 29c: Recent discovery of 400000 year old bones with mitochondrial relationship to Denisovians has raised further questions about early human emergence (Meyer et al. 2013).
Fig 29c: Recent discovery of 400000 year old bones with mitochondrial relationship to Denisovians has raised further questions about early human emergence (Meyer et al. 2013).
Some doubt has been cast on the Neanderthal interbreeding idea, attributing the effects instead to shared sequences arising from isolated African populations of the two species separating some 300-350,000 years ago, with the last exchanged genetic material some 47-65,000 years ago. However these results are contested by the original researchers from Paabo's team. They say recent analyses actually firm up the case for interbreeding. Their evidence suggests non-Africans have shared genes in common with Neanderthals for only a few tens of thousands of years, so these genes cannot predate the origin of Neanderthals.(Marshall 2012).
In 2014 more accurate datings of the demise of Neanderthals in Europe suggests they were already in serious decline 50,000 years ago probably as a result of a climate cooling phase and that by the time sapiens arrived they were already only a small fragile remnant population in scattered isolated bands (Brahic 2014, Benazzi 2011). By 39,000 years ago they had largely vanished. This doesn't imply they were actively killed off by Homo sapiens but that their territory and resources were compromised by a new invasive species. Neither is it clear that they were manifestly intellectually inferior to modern humans, as artifacts from both species show similar innovations (Barras 2014).
Genetic analysis of a bone fragment from Denisova Cave shows that it comes from an individual who had a Neanderthal mother and a Denisovan father (Slon V et al. 2018 The genome of the offspring of a Neanderthal mother and a Denisovan father Nature doi:10.1038/s41586-018-0455-x). Neanderthals and Denisovans separated from each other more than 390,000 years ago. The father's genome came from a population related to a later Denisovan found in the cave, although it also bears traces of Neanderthal ancestry. The mother came from a population more closely related to Neanderthals who lived later in Europe than to an earlier Neanderthal found in Denisova Cave, suggesting that migrations of Neanderthals between eastern and western Eurasia occurred sometime after 120,000 years ago. The finding of a first-generation Neanderthal–Denisovan offspring among the small number of archaic specimens sequenced to date suggests that mixing between Late Pleistocene hominin groups was common when they met.
 Evidence from the DNA traces left by Denisovans shows they lived on the Tibetan plateau, probably travelled to the Philippines and Laos in south Asia and might have made their way to northern China more than 100,000 years ago. They also interbred with modern humans. Their DNA, which was first found in samples from the Denisova cave in Siberia in 2010, provides most of our information about their existence. But recently scientists have pinpointed a strong candidate for the species to which the Denisovans might have belonged. This is Homo longi – or "Dragon man" – from Harbin in north-east China. This key fossil is made up of an almost complete skull with a braincase as big as a modern human's and a flat face with delicate cheekbones. Dating suggests it is at least 150,000 years old characterised by a broad nose, thick brow ridges over its eyes and large tooth sockets. Scientists in Tibet have discovered a Denisovan gene in local people, the result of interbreeding between the two species in the distant past. Crucially, this gene has been shown to help modern men and women survive at high altitudes. Evidence to support the Denisovan-Homo longi link has also been traced to the Tibetan plateau, where scientists began studying a jawbone initially found in a remote cave 3,000 metres (10,000ft) above sea level. When researchers began to study the cave where the jawbone had been originally discovered did they find its sediments were rich in Denisovan DNA and the fossil itself contained proteins that indicated Denisovan origins. Equally intriguing was the fact that the jawbone has teeth that are similar to the teeth found in Homo longi. Some were hot and low-lying, others were cold and mountainous. They represented very diverse habitats, from the Tibetan plateau to islands like Sulawesi in Indonesia] By contrast, the Neanderthals, the third large grouping of humans that evolved over the past few hundreds of thousands of years, confined themselves to the cooler climates of a region that stretched east from Europe to southern Siberia. There is good evidence that some modern humans interbred with genetically distinct Denisovans on multiple occasions. This suggests that the two groups coexisted for an extended time, with some studies suggesting a last contact as recently as 25,000 years ago..
Evidence from the DNA traces left by Denisovans shows they lived on the Tibetan plateau, probably travelled to the Philippines and Laos in south Asia and might have made their way to northern China more than 100,000 years ago. They also interbred with modern humans. Their DNA, which was first found in samples from the Denisova cave in Siberia in 2010, provides most of our information about their existence. But recently scientists have pinpointed a strong candidate for the species to which the Denisovans might have belonged. This is Homo longi – or "Dragon man" – from Harbin in north-east China. This key fossil is made up of an almost complete skull with a braincase as big as a modern human's and a flat face with delicate cheekbones. Dating suggests it is at least 150,000 years old characterised by a broad nose, thick brow ridges over its eyes and large tooth sockets. Scientists in Tibet have discovered a Denisovan gene in local people, the result of interbreeding between the two species in the distant past. Crucially, this gene has been shown to help modern men and women survive at high altitudes. Evidence to support the Denisovan-Homo longi link has also been traced to the Tibetan plateau, where scientists began studying a jawbone initially found in a remote cave 3,000 metres (10,000ft) above sea level. When researchers began to study the cave where the jawbone had been originally discovered did they find its sediments were rich in Denisovan DNA and the fossil itself contained proteins that indicated Denisovan origins. Equally intriguing was the fact that the jawbone has teeth that are similar to the teeth found in Homo longi. Some were hot and low-lying, others were cold and mountainous. They represented very diverse habitats, from the Tibetan plateau to islands like Sulawesi in Indonesia] By contrast, the Neanderthals, the third large grouping of humans that evolved over the past few hundreds of thousands of years, confined themselves to the cooler climates of a region that stretched east from Europe to southern Siberia. There is good evidence that some modern humans interbred with genetically distinct Denisovans on multiple occasions. This suggests that the two groups coexisted for an extended time, with some studies suggesting a last contact as recently as 25,000 years ago..
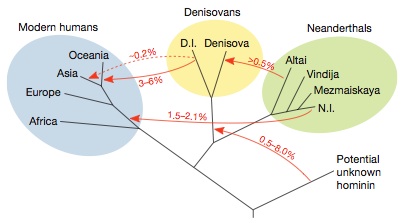
 Fig 29d: Left: A more complete picture of introgression of genes from one hominin species to another has been developed as a result of further sequencing of a slightly older Neanderthal toe bone also from Denisova cave and the analysis of the Denisovan sequence implying gene flow from an older hominin, possibly erectus or heidelbergensis. Notably the Neanderthal female was highly inbred corresponding to being the offspring of half-siblings with a common mother (Prüfer et al 2013). Right: Low heterozygosity of Neanderthal and Denisovan genomes suggests inbreeding (Prüfer et al 2017). Note also the reduced heterozygosity of non-Africans due to both Neanderthal introgression and a migratory bottleneck..
Fig 29d: Left: A more complete picture of introgression of genes from one hominin species to another has been developed as a result of further sequencing of a slightly older Neanderthal toe bone also from Denisova cave and the analysis of the Denisovan sequence implying gene flow from an older hominin, possibly erectus or heidelbergensis. Notably the Neanderthal female was highly inbred corresponding to being the offspring of half-siblings with a common mother (Prüfer et al 2013). Right: Low heterozygosity of Neanderthal and Denisovan genomes suggests inbreeding (Prüfer et al 2017). Note also the reduced heterozygosity of non-Africans due to both Neanderthal introgression and a migratory bottleneck..
There is also suggestive evidence for up to 2% interbreeding with another hominin species, in ancient African populations of Biaka Pygmies and San Bushmen (Kaplan 2011, Hammer, et al. 2011) although this is based only on statistical divergences in some loci, and lacks a sister species reference sequence, it suggests interbreeding around 35,000 years ago with a population that originally diverged from the Homo sapiens line some 700,000 years before. A further introgression of one, or more unknown hominins possibly erectus has been found in the genomes of Andaman Islanders (Mondal M et al. 2016).
 Careful analysis of Neanderthal genes on African populations, which were previously thought not to contain Neanderthal introgreesions by Joshua Akey and his team, has been found to contain a Neanderthal component around a third of the size of that in non-African populations of 1-2%, consistent with the conclusion that a wave of modern humans departed Africa some 200,000 years ago and that these people interbred with Neanderthals (Chen L et al. 2020 Identifying and Interpreting Apparent Neanderthal Ancestry in African Individuals Cell doi:10.1016/j.cell.2020.01.012.). People living somewhere in western Eurasia then moved back to Africa and interbred with people whose ancestors never left. Many models tracing Neanderthal interbreeding use what's known as a reference population -- the genomes from a group, usually from Africa, that's assumed to not have DNA from these ancient hominins. Instead, the team used large datasets to examine the probability of a particular site in the genome being inherited from Neanderthals. They tested the method with the genomes of 2,504 individuals from around the world -- East Asians, Europeans, South Asians, Americans, and largely northern Africans -- collected as part of the 1000 Genomes project. They then compared this DNA with a Neanderthal genome. Gene flow went both directions. Some of the sequences that we call Neanderthal in modern humans are actually modern human sequence in the Neanderthal genome. The new method also reveals slightly more Neanderthal DNA in modern Europeans that was previously overlooked, narrowing the 20 percent gap once thought to exist between Neanderthal ancestry in Europeans and East Asians to 8 percent or less.
Careful analysis of Neanderthal genes on African populations, which were previously thought not to contain Neanderthal introgreesions by Joshua Akey and his team, has been found to contain a Neanderthal component around a third of the size of that in non-African populations of 1-2%, consistent with the conclusion that a wave of modern humans departed Africa some 200,000 years ago and that these people interbred with Neanderthals (Chen L et al. 2020 Identifying and Interpreting Apparent Neanderthal Ancestry in African Individuals Cell doi:10.1016/j.cell.2020.01.012.). People living somewhere in western Eurasia then moved back to Africa and interbred with people whose ancestors never left. Many models tracing Neanderthal interbreeding use what's known as a reference population -- the genomes from a group, usually from Africa, that's assumed to not have DNA from these ancient hominins. Instead, the team used large datasets to examine the probability of a particular site in the genome being inherited from Neanderthals. They tested the method with the genomes of 2,504 individuals from around the world -- East Asians, Europeans, South Asians, Americans, and largely northern Africans -- collected as part of the 1000 Genomes project. They then compared this DNA with a Neanderthal genome. Gene flow went both directions. Some of the sequences that we call Neanderthal in modern humans are actually modern human sequence in the Neanderthal genome. The new method also reveals slightly more Neanderthal DNA in modern Europeans that was previously overlooked, narrowing the 20 percent gap once thought to exist between Neanderthal ancestry in Europeans and East Asians to 8 percent or less.

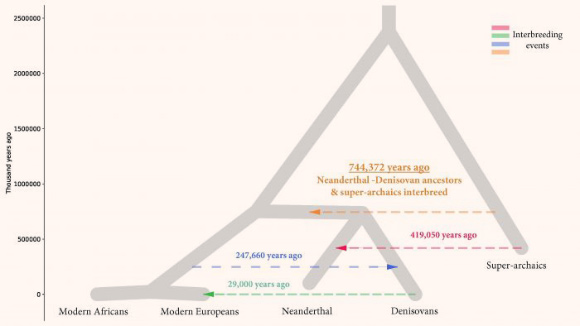
Left: African Ghost population: (A) Basic demographic model. W Afr, West Africans; Eur, European; N, Neanderthal; D, Denisovan; UA, unknown archaic. (B) Newly proposed model involving introgression into the modern human ancestor from an unknown hominin that separated from the human ancestor before the split of modern humans and the ancestors of Neanderthals and Denisovans. Below, we show the CSFS fit from the proposed model captures the U-shape observed in the data. Right: Eurasian superarchaic population and introgression events.
An "archaic ghost population" appears to have diverged from modern humans before Neanderthals split off from the family tree and left its trace in modern humans. The split appear to have taken place between 360,000 and a million years ago. These ancient humans reproduced with the ancestors of present-day Africans, just as Neanderthals reproduced with the ancestors of modern Europeans. Using a method that can identify segments of archaic ancestry without the need for reference archaic genomes, the researchers built genome-wide maps of archaic ancestry in the Yoruba and the Mende populations. DNA from this archaic population makes up between 2% and 19% of modern West Africans' genetic ancestry (Durvasula A, Sankararaman S 2020 Recovering signals of ghost archaic introgression in African populations Sci. Adv. 6 eaax5097).
The ancestors of Neanderthals and Denisovans also interbred with their own Eurasian predecessors -- members of a "superarchaic" population that separated from other humans about 2 million years ago. The superarchaic population was large, with an effective size between 20 and 50 thousand individuals (Rogers A, Harris N, Achenbach A (2020) Neanderthal-Denisovan ancestors interbred with a distantly related hominin Sci. Adv. 6 eaay5483 doi:10.1126/sciadv.aay5483).
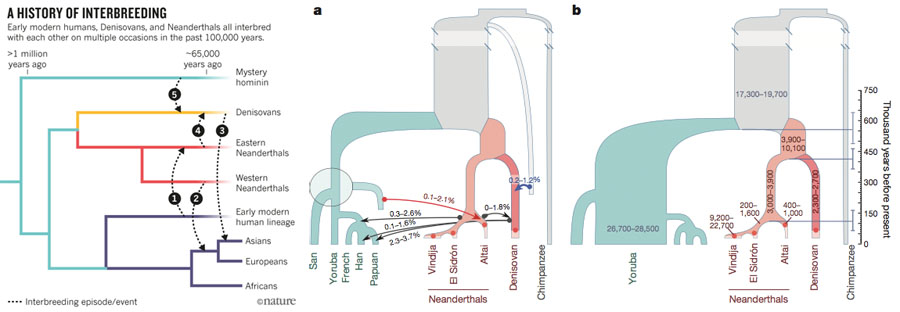
Fig 29e; Left: Earlier interbreeding also occurred 100,000 years ago in which human genes and those of another mystery hominin were transferred to Eastern Neanderthals and Denisovans respectvely (Callaway 2016). Centre and right: The same events giving percentages and populations sizes (Kuhlwilm M et al. 2016 Ancient gene flow from early modern humans into Eastern Neanderthals Nature doi:10.1038/nature16544).
The female Neanderthal sequenced in the above study also had extensive inbreeding (Marshall 2013), with up to an eighth of the genome devoid of alelle variation implying breeding between half siblings, possibly as a consequence of small isolated populations. This led to notable incidence of deformities. Those he studied have a range of deformities, many of which are rare in modern humans (Wu et al. 2013). Our genomes likewise still carry traces of past small population bottlenecks. A 2010 study concluded that our ancestors 1.2 million years ago had a population of just 18,500 individuals, spread over a vast area (Huff et al. 2010).
 Fig 29f: Relationships of intestinal bacteria in the evolution of great apes and Homo sapiens. Change in the microbiome was slow and clock-like during African ape diversification, but human microbiomes have deviated from the ancestral state at an accelerated rate. Human microbiomes have lost ancestral microbial diversity while becoming adapted for animal-based diets. (Moeller et al. 2014).
Fig 29f: Relationships of intestinal bacteria in the evolution of great apes and Homo sapiens. Change in the microbiome was slow and clock-like during African ape diversification, but human microbiomes have deviated from the ancestral state at an accelerated rate. Human microbiomes have lost ancestral microbial diversity while becoming adapted for animal-based diets. (Moeller et al. 2014).
Chromosomes contain a variety of markers that can be used to compare diverse populations and infer an evolutionary relationship between them. These include the slowly varying protein polymorphisms of coding regions which are useful for long-term trends, single nucleotide polymorphisms, and non-coding region changes (mutation rates about 2.5 x 10-8 per base pair per generation and useful for reconstructing evolutionary history only over millions of years) insertion and deletion events (about 8% of polymorphisms, extending from one to millions of nucleotides), particularly those driven by transposable elements such as the LINEs and even more frequent SINEs, non-coding micro-satellites (mutation rate 10-5 - 10-2 due to repeat slippage) and mini-satellite regions of repeating DNA (mutation rates as high as 2 x 10-1 due to meiotic recombination in sperm) that both evolve rapidly and are not subject to the strong selection of coding regions which can differentiate changes over the much shorter time scales of modern human migration.
The insertions and deletions of the million or so Alu elements in the human genome are particularly useful, as the most active sub-population of about 1000 Alu is actively transcribing and undergoing rapid change. A subpopulation of Alu are capable of generating new coding regions (exons), when inserted into non-coding introns between spliced sections of a translated mRNA, because one base-pair change within Alu leads to formation of a new exon reading into the surrounding DNA. This is not necessarily deleterious because alternative splicing still allows the original protein to be made as well. We have the highest number of introns per gene of any organism, and thus have to have gained an advantage from this costly error-prone process. Alus may have given rise, through alternative splicing, to new proteins that drove primates' divergence from other  mammals. Recent studies have shown that the nearly identical genes of humans and chimps produce essentially the same proteins in most tissues, except in parts of the brain, where certain human genes are more active and others generate significantly different proteins through alternative splicing of gene transcripts. Our divergence from other primates may thus be due in part to alternative splicing.
mammals. Recent studies have shown that the nearly identical genes of humans and chimps produce essentially the same proteins in most tissues, except in parts of the brain, where certain human genes are more active and others generate significantly different proteins through alternative splicing of gene transcripts. Our divergence from other primates may thus be due in part to alternative splicing.
Fig 29g: Left: Southern African ancient genomes estimate modern human divergence to 350,000 years ago, with a genetic split between the Khoisan and other Africans 260,000 years ago, shortly after humankind's origins and around the time of the Florisbad individual. Khoisan people then diverged into two genetically distinct populations around 200,000 years ago. Right: Population movements deduced from ancient DNA (Soglund et al. 2017). There has also been an admixing of Western Eurasian non-African genomes firstly into Ethiopia 2,700–3,300 years ago and eventually down ino the Khoe San between 1,800 and 900 years ago, where Neanderthal genes due to the Eurasian Neanderthall introgression showed up in a genetic assay (Pickrell et al. (2014). This has historical consistency with cultural accounts of Makeda, the Ethopian tale of the Queen of Sheba .
An in depth study into human origins (Schlebusch et al 2012, 2017) estimates, using ancient and modern genome data, including the benchmark genome of a boy from Southern Africa 2000 years ago, found that Homo sapiens originated as a genetically distinct species between 350,000 and 260,000 years ago. African populations branch in two directions and then further subdivide around 200,000 years ago. Non-African populations, appeared shortly after 100,000 years ago. A genetic split between the Khoisan and other Africans occurred roughly 260,000 years ago, shortly after humankind's origins and around the time of the Florisbad individual. Khoisan people then diverged into two genetically distinct populations around 200,000 years ago, the researchers calculate.

L0 phylogenetic tree, geographical distributions of the major southern African L0 haplogroup and out-of-homeland L0 dispersal routes.
Scientists in 2019 (Chan E et al. 2019) have traced the ancestral home region of all living humans to a vast wetland that sprawled over much of modern day Botswana and served as an oasis in an otherwise parched expanse of Africa. The swathe of land south of the Zambezi River became a thriving home to Homo sapiens 200,000 years ago, the researchers suggest, and sustained an isolated, founder population of modern humans for at least 70,000 years. The group remained in the region until a shift in the climate, driven by changes in the Earth's tilt and orbit, brought rains to the north-east and south-west, producing lush green corridors that allowed the early humans to spread into new territories, the scientists say. The conclusions are based on an analysis of 1,217 samples of mitochondrial DNA from people living in southern Africa today, including the Khoisan Geological, archaeological and fossil evidence about the climate and broader ecosystem in the region at the time indicates that a body of water the size of New Zealand, called Lake Makgadikgadi, once dominated the area, but had started to break up into a massive wetland 200,000 years ago. It would have been very lush and it would have provided a suitable habitat for modern humans and wildlife to have lived. According to the DNA analyses, the L0 lineage split 130,000 years ago when some of the founder population moved north-east along a green vegetated route that opened up as rains drenched the arid land. A second wave of migration headed south-west about 20,000 years later as rainfall also increased vegetation in that direction. Those who headed north-east gave rise to farming populations, while those who went south became coastal foragers. However, analyses of the male-inherited Y chromosome suggest our Y-carrying ancestors may have originated from west Africa. Further studies, which have looked at whole genomes, point to populations who migrated out of Africa originating in the continent's east. These and many other data suggest that we are an amalgam of ancestry from different regions of Africa with, of course, the addition of interbreeding from other human groups outside the continent.
A 2017 study of ancient genomes (Skoglund et al. 2017) coaxed DNA from the remains of 15 ancient sub-Saharan Africans, from a variety of geographic regions dating from about 500 to 8500 years old. The researchers compared these, along with one other ancient genome from the region, against those of nearly 600 present-day people from 59 African populations and 300 people from 142 non-African groups. The results indicate that there was a complete population replacement eliminating the existing gatherer-hunter population realted to the Khoe-San (yellow) as farmers moved into Malawi, unlike the picture in Europe where famers admixed witht eh existing gatherer-hunter population. The Hadza (red) were found to have had a central position in ancient african migration and may have also participated in the out of Africa migration. The study also found that West Africans (green) can trace their lineage back to a human ancestor that may have split off from other African populations even earlier than the Khoe-San. A missing link with a Near Eastern herder population that returned to Africa. Adaptions were found for resisting UV radiation (e.g. in the Kalahari, and for bitter tastes providing better capacity to tell poisonous plants.
Unlike the inbred genetic profile of the Altai Neanderthals (Prüfer et al 2013) and those from Vindija Cave in Croatia (Prüfer et al 2017), genetic sequencing of ancient individuals the Sungir group of upper paleolithic Homo sapiens remains (Sikora et al. 2017) dating from around 34,000 years ago, shows they lived in groups with few close relatives, thus limiting inbreeding. The researchers suggest this both provided enhanced genetic fitness and also catalysed social networking conducive to social sophistication unique to Homo sapiens and similar to modern gatherer-hunter groups with exogamous mating patterns. The site is one of the earliest examples of ritual burials and constitutes important evidence of the antiquity of human religious practices.
If we consider the likely effects of the out of Africa hypothesis, we would expect that founding African populations not subject to active expansion and migration would have greater genetic diversity and that the genetic makeup of other world populations would come from a subset of the African diversity, consisting of those subgroups who migrated. This picture is complicated by the evidence for one or more bottlenecks that reduced the genetic diversity of the surviving human population to 3000-10,000 breeding pairs around 70,000 years ago, which has been associated with the supervolcanic Toba eruption in Sumatra.
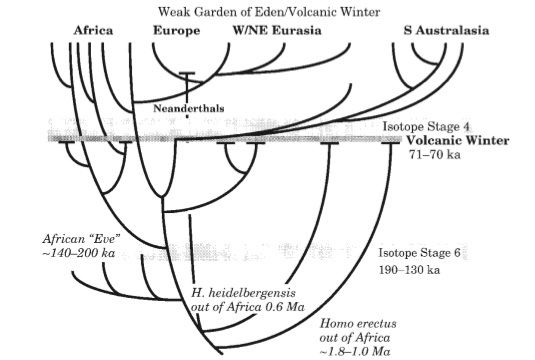 Fig 29h: The Volcanic Winter/Weak Garden of Eden model proposed in Ambrose 1998. Population subdivision due to dispersal within African and to other continents during the early Late Pleistocene is followed by bottlenecks caused by volcanic winter, resulting from the eruption of Toba, around 71,000 years ago. The bottleneck may have lasted either 1000 years, during the hyper-cold stadial period between Dansgaard-Oeschlger events 19 and 20, or 10,000 years, during oxygen isotope stage 4. Population bottlenecks and releases are both synchronous. More individuals survived in Africa because tropical refugia were largest there, resulting in greater genetic diversity in Africa. Research in 2018 has established that Humans thrived in southern Africa throughout the period (Smith et al. 2018).
Fig 29h: The Volcanic Winter/Weak Garden of Eden model proposed in Ambrose 1998. Population subdivision due to dispersal within African and to other continents during the early Late Pleistocene is followed by bottlenecks caused by volcanic winter, resulting from the eruption of Toba, around 71,000 years ago. The bottleneck may have lasted either 1000 years, during the hyper-cold stadial period between Dansgaard-Oeschlger events 19 and 20, or 10,000 years, during oxygen isotope stage 4. Population bottlenecks and releases are both synchronous. More individuals survived in Africa because tropical refugia were largest there, resulting in greater genetic diversity in Africa. Research in 2018 has established that Humans thrived in southern Africa throughout the period (Smith et al. 2018).
In the case of mitochondrial mtDNA (mutation rate about 2.5 x 10-7) and its hyper-variable D-loop (mutation rates as high as 4 x 10-3), which is transmitted only down the maternal line (see Tishkoff and Verrelli for caveat) and the non-recombining majority of the Y-chromosome which is transmitted only down the paternal line, each with no recombination, we would expect greater diversity going deeper into the historical tree of divergence, with certain existing groups who have retained the founding patterns of survival and have not undergone rapid population expansions to retain an increasingly diverse source variation. All these features are broadly observed in the genetic data to date.
 Fig 30: (a) MtDNA tree for African groups showing haplotypes of !Kung, Mbuti and Biaka as well as the line coming out of Africa (Chen et. al.). (b) Diagram of world migration and regional differentiation of successive mtDNA haplotypes (Gilbert). (c) mtDNA distances between founding African groups including Hadza (clicks) Khwe is from (Knight et. al.). Recent mtDNA evidence suggests a first wave of migration down the coast of Asia all the way to Australia (Forster et. al.).
Fig 30: (a) MtDNA tree for African groups showing haplotypes of !Kung, Mbuti and Biaka as well as the line coming out of Africa (Chen et. al.). (b) Diagram of world migration and regional differentiation of successive mtDNA haplotypes (Gilbert). (c) mtDNA distances between founding African groups including Hadza (clicks) Khwe is from (Knight et. al.). Recent mtDNA evidence suggests a first wave of migration down the coast of Asia all the way to Australia (Forster et. al.).
Most studies of non-coding regions of autosomal, X-chromosome, and mitochondrial mtDNA genetic variation (which are desirable markers because they are not so subject to selection and thus have relatively neutral drift) show higher levels of genetic variation in African populations compared to non-African populations, using many types of markers. Although some studies of Y-chromosome variation have observed higher heterozygosity levels in non-African populations, the African populations have higher levels of pairwise sequence differences, consistent with these populations being ancestral. High levels of diversity in African populations alone do not prove that African populations are ancestral. A recent bottleneck event and/or colonization and extinction events among non-African populations, or a more recent onset of population growth in non-Africans, could also cause a decrease in genetic diversity (Tishkoff and Verrelli). In fact the complete inter-fertility of all human populations and the relative lack of genetic divergence by comparison with the few remaining chimp colonies in the wild (Hrdy 183) does indicate a significant bottleneck. The genetic data is consistent with a human emergence from a population of only 10,000 around 100,000 years ago. This is also consistent with the delayed maturation, long birth spacings as a result of prolonged lactation and high infant mortality seen in gather-hunter populations such as the !Kung. At such low growth rates a population of 100 would take 50,000 years to reach 10,000 (Hrdy 183).
 Fig 31: Patterns of male migration. The Genographic Project - a partnership between National Geographic and IBM - will collect DNA samples from over 100,000 people worldwide to provide a high-resolution genetic map of human migration.
Fig 31: Patterns of male migration. The Genographic Project - a partnership between National Geographic and IBM - will collect DNA samples from over 100,000 people worldwide to provide a high-resolution genetic map of human migration.
However studies of protein polymorphisms as well as mtDNA haplotypes, X-chromosome and Y-chromosome haplotypes, autosomal microsatellites and minisatellites, Alu elements, and autosomal haplotypes indicate that the roots of the population trees constructed from these data are composed of African populations and/or that Africans have the most divergent lineages, as expected under a recent African origin rather than a multi-regional emergence model. Additionally, studies of autosomal, X-chromosomal haplotype and mtDNA variation indicate that Africans have the largest number of population-specific alleles and that non-African populations harbor a subset of the genetic diversity that is present in Africa, as expected if there was a genetic bottleneck when modern humans migrated out of Africa. Analysis of genetic variation among ethnically diverse human populations indicates that populations cluster by geographic region (i.e., Africa, Europe/Middle East, Asia, Oceania, New World) and that African populations are highly divergent. The mtDNA studies hypothesize a primal female ancestor - the African Eve - around 150,000 years ago (Chen et. al.) while the Y-chromosome Adam is more recent, at around 90,000 years ago (Underhill et. al.) consistent with the greater reproductive variance of males than females. Differences between the Y- and mtDNA distributions indicate how migration, intermarriage and female exogamy have affected the gene pool. The genetic patterns of both these and autosomal microsatellites (Zhivotovsky et. al.) are consistent with founding African diversity with migratory radiations to form other world populations, with deep founding radiations to the forest people such as the Biaka and Mbuti, Khoisan click-language speaking !Kung-san bushmen of Botswana and the Sandawe of Tanzania, and possibly the Hadzabe, as well as the forest people such as the Mbuti and Biaka 'pygmies' who have adopted the Bantu languages of the farming neighbours with which they now share semi-symbiotic relationships. Along with some Ethiopian and Sudanese sub-populations, these groups may represent some of the oldest and deeply diversified branches of modern humans.
 Fig 32: (Right) Genographic project study of mitochondiral origins shows a deep split separating Khoisan mitochondrial inheritance from other groups, including those migrating out of Africa, suggesting a separation of some 100,000 years possibly caused by long term drought in Africa (Behar et al.) (Left) Phylogeny of 526 complete mitochondrial genomes depicting the earliest diverged modern human maternal lineages, including the first ancient Khoesan mtDNA (StHe) within the L0d2c lineage. All non-L0d2c genomes have been collapsed with each triangle representing the relative diversity of the corresponding haplogroups and subclades. In 2014 the skeleton of a male marine forager discovered at St. Helena, a carbon dated to 2,330 ± 25 years before the present, displays one of the oldest mitochondrial clades L0d2c1c, unlike its Khoe-language based sister-clades (L0d2c1a and L0d2c1b) most closely related to contemporary indigenous San-speakers (specifically Ju). whose ancestors diverged from other humans roughly 150,000 years ago. (Morris et al. 2014) before Khoekhoe speaking pastoralists arrived 500 years later.
Fig 32: (Right) Genographic project study of mitochondiral origins shows a deep split separating Khoisan mitochondrial inheritance from other groups, including those migrating out of Africa, suggesting a separation of some 100,000 years possibly caused by long term drought in Africa (Behar et al.) (Left) Phylogeny of 526 complete mitochondrial genomes depicting the earliest diverged modern human maternal lineages, including the first ancient Khoesan mtDNA (StHe) within the L0d2c lineage. All non-L0d2c genomes have been collapsed with each triangle representing the relative diversity of the corresponding haplogroups and subclades. In 2014 the skeleton of a male marine forager discovered at St. Helena, a carbon dated to 2,330 ± 25 years before the present, displays one of the oldest mitochondrial clades L0d2c1c, unlike its Khoe-language based sister-clades (L0d2c1a and L0d2c1b) most closely related to contemporary indigenous San-speakers (specifically Ju). whose ancestors diverged from other humans roughly 150,000 years ago. (Morris et al. 2014) before Khoekhoe speaking pastoralists arrived 500 years later.
Such recent genetic evidence has laid bare the relationships between some of the founding human groups spread across Africa from the 'Cushite' horn of Ethiopia to the southern Kalahari. Mitochondrial DNA studies have highlighted the ancient origin of the !Kung San and of pygmy peoples of the Congo Basin such as the Mbuti and the Biaka.
 Fig32b: The largest ever study of global genetic variation in the human Y chromosome has elucidated the phylogenetic tree of men. Some parts of the tree were more like a bush, with many branches originating at the same point indicating there was an explosive increase in the number of men carrying that type of Y chromosome The earliest occurred 50,000-55,000 years ago, across Asia and Europe, and 15,000 years ago in the Americas. There were also later expansions in sub-Saharan Africa, Western Europe, South Asia and East Asia, between 8,000 and 4,000 years ago. The earlier population increases probably resulted from the first peopling by modern humans of vast continents, where plenty of resources were available and the later from advances in technology that could be controlled by small groups of men: Wheeled transport, metal working and organized warfare (Poznik, G et al. 2016)
Fig32b: The largest ever study of global genetic variation in the human Y chromosome has elucidated the phylogenetic tree of men. Some parts of the tree were more like a bush, with many branches originating at the same point indicating there was an explosive increase in the number of men carrying that type of Y chromosome The earliest occurred 50,000-55,000 years ago, across Asia and Europe, and 15,000 years ago in the Americas. There were also later expansions in sub-Saharan Africa, Western Europe, South Asia and East Asia, between 8,000 and 4,000 years ago. The earlier population increases probably resulted from the first peopling by modern humans of vast continents, where plenty of resources were available and the later from advances in technology that could be controlled by small groups of men: Wheeled transport, metal working and organized warfare (Poznik, G et al. 2016)
Y-chromosome studies have shown the !Kung share a most ancient haplotype with sub-populations from Ethiopia and the Sudan. According to an overall survey of genetic research by Sarah Tishkoff of theUniversity of Maryland, the most deeply ancestral known human DNA lineages may be those of East Africans, such as the Sandawe, who share many phenotypic features and a click language with the !Kung.  This suggests southern Khoisan-speaking peoples originated in East Africa. The most ancient populations are now believed to also include the Sandawe, Burunge, Gorowaa and Datog people of Tanzania. The Burunge and Gorowaa migrated to Tanzania from Ethiopia within the last 5,000 years consistent with an ancient founding population in this area. Echoes of the earliest language spoken by ancient humans tens of thousands of years ago may have been preserved in the distinctive clicking sounds still spoken by some existing African tribes.
This suggests southern Khoisan-speaking peoples originated in East Africa. The most ancient populations are now believed to also include the Sandawe, Burunge, Gorowaa and Datog people of Tanzania. The Burunge and Gorowaa migrated to Tanzania from Ethiopia within the last 5,000 years consistent with an ancient founding population in this area. Echoes of the earliest language spoken by ancient humans tens of thousands of years ago may have been preserved in the distinctive clicking sounds still spoken by some existing African tribes.
(Above) Baka Adam vs Mbuti Eve. The new data from Poznik et al (2013) does not necessarily mean Eve is exactly the same age as Adam. (Below) New resolution of the Y-spread immediately out of Africa.
Highlighting unique features of human genetic evolution, are two key genes whose mutations cause microcephaly, consistent with increased brain size, whose rapid spread through the human population may coincide with spurts in human culture. Microcephalin (Evans et. al.) appeared ~37,000 years ago coinciding with the birth of culture and ASPM spread from the Near East around 5000 years ago (Mekel-Bobrov et. al.). However studies linking these variants have failed to find differences in intelligence and results remain highly controversial (Balter 2006). Nevertheless, these results are consistent with an overall examination of linkage disequilibrium in single nucleotide polymorphisms (Ding et. al.) which indicate that about 7% of our genes have been subject to selection in the last 50,000 years, a figure similar to domestication of maize, including genes for protein metabolism, disease resistance and brain function.

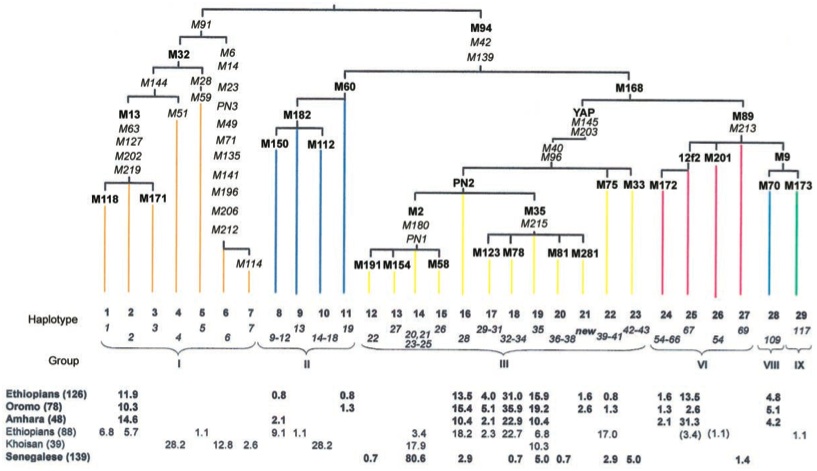
Fig 33: Left: (a) Non-recombining Y-chromosome evolutionary tree (Underhill et. al.) (b) Geographical distribution showing the ancient haplotype shared by the San and Ethiopian and Sudanese sub-populations. (c) Genetic distances between Khoisan and forest peoples sharing M112 a Y-chromosome allele common only in these groups showing great genetic distance between Hadzabe and San peoples (Knight et. al.) . (d) Autosome satellite analysis confirming ancient divergence of San and forest peoples leading to migration from Africa (Zhivotovsky et. al.). Right: The genetic structure of 126 Ethiopian and 139 Senegalese Y chromosomes was investigated by a hierarchical analysis of 30 diagnostic biallelic markers selected from the worldwide Y-chromosome genealogy. The present study reveals that only the Ethiopians share with the Khoisan the deepest human Y-chromosome clades. This confirms the ancestral affinity between the Ethiopians and the Khoisan, which has previously been suggested by both archaeological and genetic findings (Semino et al.).
 Y-chromosome studies have shown the !Kung share a most ancient haplotype with sub-populations from Ethiopia and the Sudan, suggesting they are parts of an ancient widespread population later divided by the Bantu expansion. According to an overall survey of genetic research by Sarah Tishkoff of the University of Maryland, the most deeply ancestral known human DNA lineages may be those of East Africans, such as the Sandawe, who share many phenotypic features and a click language with the !Kung. This suggests southern Khoisan-speaking peoples originated in East Africa. The most ancient populations are now believed to also include the Sandawe, Burunge, Gorowaa and Datog people of Tanzania. The Burunge and Gorowaa migrated to Tanzania from Ethiopia within the last 5,000 years consistent with an ancient founding population in this area. Echoes of the earliest language spoken by ancient humans tens of thousands of years ago may have been preserved in the distinctive clicking sounds still spoken by some existing African tribes.
Y-chromosome studies have shown the !Kung share a most ancient haplotype with sub-populations from Ethiopia and the Sudan, suggesting they are parts of an ancient widespread population later divided by the Bantu expansion. According to an overall survey of genetic research by Sarah Tishkoff of the University of Maryland, the most deeply ancestral known human DNA lineages may be those of East Africans, such as the Sandawe, who share many phenotypic features and a click language with the !Kung. This suggests southern Khoisan-speaking peoples originated in East Africa. The most ancient populations are now believed to also include the Sandawe, Burunge, Gorowaa and Datog people of Tanzania. The Burunge and Gorowaa migrated to Tanzania from Ethiopia within the last 5,000 years consistent with an ancient founding population in this area. Echoes of the earliest language spoken by ancient humans tens of thousands of years ago may have been preserved in the distinctive clicking sounds still spoken by some existing African tribes.
Fig 34: Human divergence trees calculated by single nucleotide polymorphisms (SNPs) top left (Li et. al.) bottom right (Jakobsson et. al.). Trees for haplotypes and copy number variation between populations (Li et. al.). (click to enlarge).
 In a counterpoint to these studies, (Hein, Rohde et. al.) estimate that the repeated spreading of family trees by sexually recombining mobile populations and differences in reproductive rates leads to an estimate of the most recent common ancestor of our global populations existing just 3,500 years ago, excepting these most isolated groups.
In a counterpoint to these studies, (Hein, Rohde et. al.) estimate that the repeated spreading of family trees by sexually recombining mobile populations and differences in reproductive rates leads to an estimate of the most recent common ancestor of our global populations existing just 3,500 years ago, excepting these most isolated groups.
Further studies of the nuclear genome, using SNPs (single nucleotide polymorphisms), CNVs (copy number variation) and haplotype have thrown up reasonably consistent maps of regional divergence of principal human groups, demonstrating correspondence to the "Out of Africa" hypothesis and consistent with major patterns of migration.
Biallellic deletion-based tree including Neanderthals and Denisovans as outliers (Sudmant et al. 2015).
In 2015 (Sudmant et al.) the study of CNVs (deletions and gene duplications) was expanded to 236 individuals from 125 distinct human populations including an in depth exploration of duplications which require more advanced techniques to assess. In total, 7.01% of the human genome is variable due to CNVs in contrast to 1.1% due to single-nucleotide variation. Deletions (loss of sequence) were less common (representing 2.77% of the genome) compared to duplications (4.4% of the genome), suggesting that many duplications are fixed because they are advantageous. CNVs mapping to segmental duplications were larger on average (median of 14.4 kbp), than CNVs mapping to the unique portions of the genome (median of 6.2 kbp).
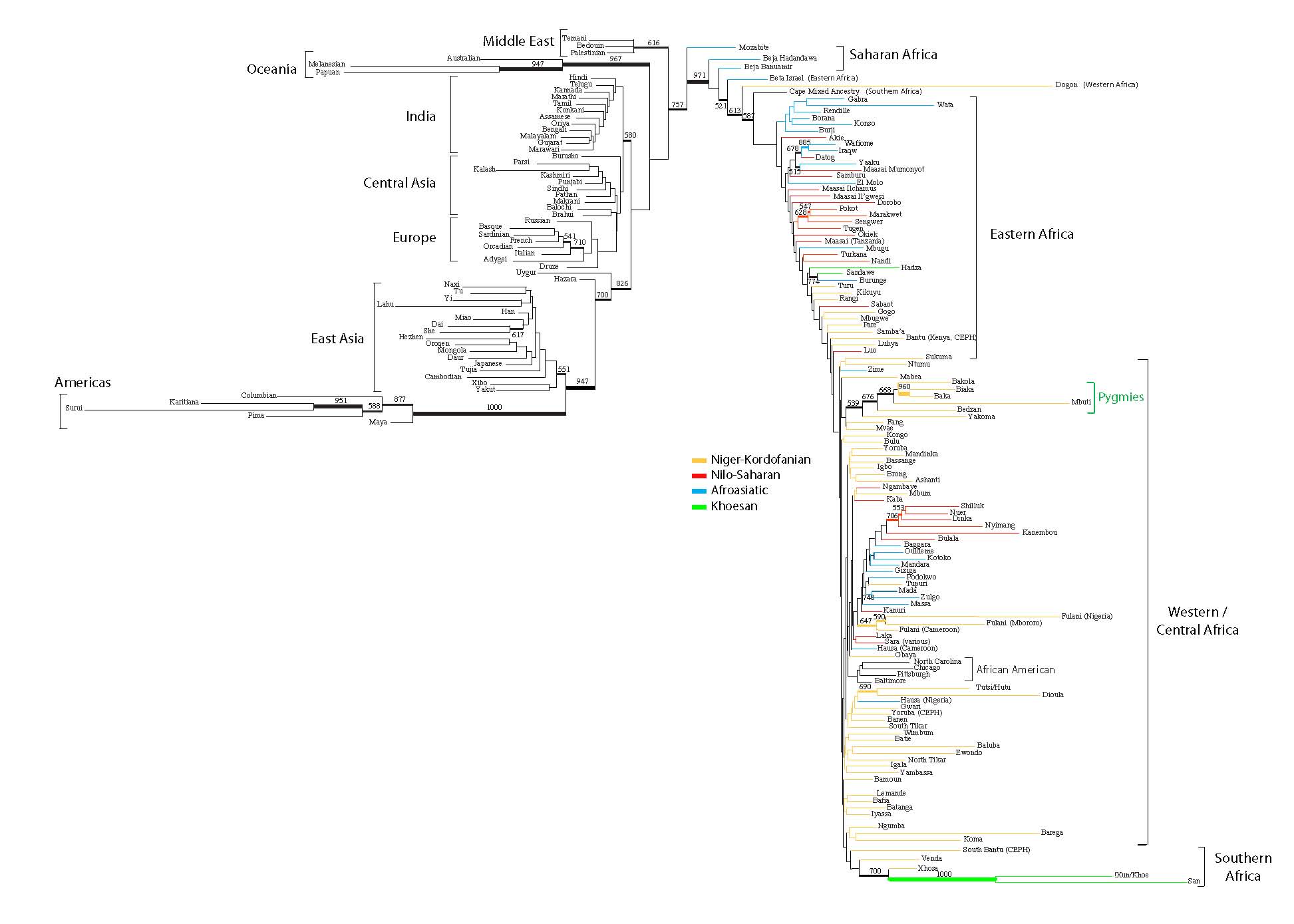

Fig 35: Left: In 2009, Tishkoff et. al. reported on a major study of African and African American evolution containing the most detailed information on African diversity to date (click to enlarge). Right: Reproductive bottleneck in Y-chromosome diversity began about 10,000 years ago and continued for several millennia (Karmin et al. 2015). Inset shows 11 independent areas of primal agriculture discovered. Evidence of animal husbandry has also been found in Turkey 10500 years ago. (The real first farmers: How agriculture was a global invention New Sci 28 Oct 2015).
In 2015, research into the comparative population diversity of maternal mitochondrial DNA and the male Y-chromosome led to an astounding contrast. Around 10,000 years ago, corresponding to the birth of agriculture, the diversity of the Y-chromosome underwent a collapse across vast areas on the human-colonized planet. There is no evidence this was a result of direct biological or genetic factors as there were no differences between differing Y-clades. The conclusion is that the effect was driven by cultural changes associated with agriculture in which powerful men were able to reproductively exploit large numbers of women and transmit their reproductive success on to their male heirs, squeezing the majority of males out of the reproductive race. Estimates of this phase of extreme reproductive polygyny suggest that for every reproducing male there were 17 reproductive females effectively making harems the predominant form of sexual relationship (Karmin et al. 2015).
A member of the research team suggested that only a few men accumulated lots of wealth and power, leaving nothing for others. These men could then pass their wealth on to their sons, perpetuating this pattern of elitist reproductive success. Then, as more thousands of years passed, the numbers of men reproducing, compared to women, rose again.
 Fig 35b: Evolutionary tree of human polygyny among ethnic groups (Minochera et al. 2018). Pathogen stress and assault frequency emerged as the predictors most strongly associated with polygyny, which had been considered evidence for female choice of good genes and male intra-sexual competition or male coercion, respectively. Mixed support was found for a polygyny threshold based on variance in male wealth.
Fig 35b: Evolutionary tree of human polygyny among ethnic groups (Minochera et al. 2018). Pathogen stress and assault frequency emerged as the predictors most strongly associated with polygyny, which had been considered evidence for female choice of good genes and male intra-sexual competition or male coercion, respectively. Mixed support was found for a polygyny threshold based on variance in male wealth.
In more recent history, as a global average, about four or five women reproduced for every one man, still a highly polygynous picture that leads into some of the great patriarchs of history from Ghengis Khan whose Y-chromosome continues to exist in 8% of men in 16 populations spanning Asia and some 0.5% of males worldwide (Zerjal, T. et al. 2003) to Udayama who was said to keep 16,000 virgins behind flaming walls (Ridley 1993, Watson 1995). Several other great founders of Y-chromosome lineages have been discovered (Callaway 2015b, Balaresque 2014).
This comes as an ironical twist since it is assumed that agriculture was an invention of women coming out of their role as gatherers in gather-hunter societies and provides a new perspective on the societies of the planter queens where female deities appear to have been worshipped at the same time as this extreme form of male reproductive elitism. The other thing that is really stunning about this effect is that it has been repeated widely across diaparate world cultures, from China through the Near East to Europe and even Precolombian America.
An explanation for this extreme genetic skewing has been proposed in terms of cultural hitch-hiking amid extreme competition between patrilineal kin groups (Zeng, Aw & Feldman 2018 Nature Comms. 9:2077 doi:10.1038/s41467-018-04375-6). The effect emerges around 10,000 years ago and continues for 5,000 years, largely predating the agrarian urban empires.
Even given excessive centralization of reproductive power from overlords, leading to the Genghis Khan effect, where due to three generations of Khan rulers establishing huge reproductive harems, 0.5% of the Y-chromosomes on the planet come from Genghis Khan and no less than 8% in areas of central Asia representing 16 million men in all (Zerjal T et al. 2003 The Genetic Legacy of the Mongols Am J Hum Genet. 72/3 717-721 doi:10.1086/367774). Nevertheless, reproductive inequality in agricultural societies is unlikely to become skewed to as high a sex ratio as 1:17.
A key proposal reinforced by dynamical systems simulations and historical analysis is that patrilineal kin groups fighting competitive battles between clans of related individuals, act to eradicate entire patriarchal genealogies from the record through lethal conflicts which annihilate an entire genetic clade of males at a single sitting. The winners can then enjoy the enhanced reproductive success of the opposing clan's women folk who in a patrilineal system have joined the group exogenously the victorious clan reaping enhanced reproductive success, while the defeated clan disappears from the record entirely. The combine effects of deletion of whole Y lineages combined with cultural hitch-hiking by taking advantage of the enlarged pool of fertile females gives a two-process explanation of how the Y-diversity can plummet while the mitochondrial diversity does not. A similar process can be found among warrior societies such as the Yanomamo.
When three populations Khoisan from Africa, Mongolian Khalks and Papua New Guinea Highlanders were examined for the differences in age between the Y-chromosome Adam and the mitochondrial Eve, the ages of all three groups had a roughly 2:1 difference in age (SAN 73.6 kya vs 176.5 kya, MNG 43.6 kya vs 134.4 kya and PNG 45.5 kya vs 81.05 kya). These results are most consistent with a higher female effective population size skewed toward an excess of females by sex-biased demographic processes. They demonstrate that overall female reproductive populations sizes throughout the last 100,000 years of human evolution have been effectively polygynous by a factor of around 2:1.
For an authoritative recent overview of the peopling of the planet in terms of genomics see Nielsen et al. (2017).
Language and Cultural Evolution


Fig 36: Left: Evolutionary tree of Indo-European languages suggests a possible radiation corresponding to the Kurgans occurred around 6,900 years ago and that they were preceded by Hittite migrations into Anatolia. Time scales in red are BP (Gray and Atkinson). Significantly Tocharian appears in Buddhist writings from China's Xinjiang province, indicating early far-eastern spread. Inset: hypothetical relationship between Indo-European and wider language groups such as Afro-Asiatic (click to enlarge). Looking at the Indo-European origin geographically Bouckaert et al. (2012) found decisive support for an Anatolian origin over a steppe origin. Both the inferred timing and root location of the Indo-European language trees fit with an agricultural expansion from Anatolia beginning 8000 to 9500 years ago. Right: The DNA analysis of widespread fossil and current genomes has led to confirmation of a great Yamnaya migration from the Steppe around 4500 years ago which almost completely replaced the gatherer-hunter populations of Europe (Haak et al. 2015). See also Allentoft et al. (2015).
The evolutionary tree of human ethnic and migratory peoples bears an interesting relationship with the corresponding tree of languages, in which language appears to have a cultural evolutionary capacity of its own occurring more rapidly than genetic evolution, complementing the biological evolution of human populations.
Counterposing the idea of a hardwired genetic basis for the human capacity for spoken language, as exemplified by Chomsky's generative grammar, is the theory of language as an evolutionary 'parasites' converging towards internal efficiency through the modularity of their grammar and word set. Darwin (1904), the founder of the evolutionary approach (1859) speculated that language was potentially an invention: "Man not only uses inarticulate cries, gestures and expressions, but has invented articulate language, if indeed the word invented can be applied to a process completed by innumerable steps half consciously made". Morten Christiansen (Christiansen and Kirby 2003) question the need to invoke a Chomskian generative grammar. Instead, they argue, language has adapted to utilize more general cognitive processing capacities that were already part of our ancestors' brains before language came along. Among these, he focuses on 'sequential learning' - the ability to encode and represent the order of the discrete elements in a sequence. This ability is not unique to humans: mountain gorillas, for example, use it in the complicated preparation of certain 'spiky' plant foods, where a sequence of tasks is required to remove the edible part. Language, he says, is a 'non-obligate mutualistic endosymbiont' - a kind of evolutionary structure like a 'symbolic virus'. Kirby suggests our brains are not so specifically designed for language and that we appear to be biologically adapted to language because language, which evolves much faster than biology has culturally adapted to us, gaining semantic power and representational efficiency as it evolves. Languages as different as Danish and Hindi have evolved in less than 5000 years from a common Proto-Indo-European ancestor. Yet it took up to 200,000 years for modern humans to evolve from archaic Homo sapiens. This tallies well with the fact that written languages cannot possibly have a hard-wired basis, having only existed for the last 4000 or so years and being a product of only a few cultures, yet we can adapt our visual pattern recognition readily to become fully literate.
Confirmation that the tree of life of language evolution is a cultural evolutionary phenomenon, rather than a cognitive universal, by implication determined genetically (Ball 2011) has come with the work of Russel Gray and coworkers (Dunn et al. 2011). In the Nature editorial "Universal Truths" the scope of this is made clear. There are two theories of language universality which delineate the field. Noam Chomsky proposed that the brain is genetically endowed with rules providing brain modules which express a universal grammar. Joshua Greenberg, takes a more empirical approach, identifying traits (particularly in word order) shared by many languages, which are considered to represent biases that result from cognitive constraints. Gray and his colleagues have put both to the test using phylogenetic methods to examine four family trees that between them represent more than 2,000 languages. They considered whether what we call prepositions occur before or after a noun ("in the boat" versus "the boat in") and how the word order of subject and object work out in either case ("I put the dog in the boat" versus "I the dog put the canoe in"). A generative grammar should show patterns of language change that are independent of the family tree or the pathway tracked through it, whereas Greenbergian universality predicts strong co-dependencies between particular types of word-order relations (and not others). Neither of these patterns is borne out by the analysis, suggesting that the structures of the languages are lineage-specific and not governed by universals.
Quentin Atkinson (2011) has taken this a step further. Human genetic and phenotypic diversity declines with distance from Africa, as predicted by a serial founder effect in which successive population bottlenecks during range expansion progressively reduce diversity, consistent with the "out of Africa" hypothesis. Likewise Atkinson showed that the number of phonemes used in a global sample of 504 languages is clinal and fits a serial founder-effect model of expansion from an inferred origin in Africa - in effect a cultural evolutionary process. In Atkinson's words this "points to parallel mechanisms shaping genetic and linguistic diversity and supports an African origin of modern human languages.
To quote Jabr (2011) "Earlier research has shown that the more people speak a language, the higher its phonemic diversity. Africa turned out to have the greatest phonemic diversity - it is the only place in the world where languages incorporate clicks of the tongue into their vocabularies, for instance - while South America and Oceania have the smallest. Remarkably, this echoes genetic analyses showing that African populations have higher genetic diversity than European, Asian and American populations.

Fig 37: Hypothetical core of all human languages from Greenhill et al. (2010) further work associated with Gray and Atkinson's research (click to enlarge). The Nature article above implies "languages evolve in their own idiosyncratic ways, rather than being governed by universal rules set down in human brain patterns".

Fig 38: Tree of world religions included to turn the tables on creationist deniers of evolution (click to enlarge). Lacks detail for African tribal religions (see Culture out of Africa).
Finally we note that, contrary to creationist and intelligent design notions of life needing a design specification from a third party "God" whith no detectable presence in the natural universe, religions themselves can be seen to show a similar form of cultural evolution to the languages they are expressed in. The evolutionary tree of life remains the root and branch from which, through the muck and slime of sexual recombination, human intelligence, culture and religion has sprung. Thus evolution is fecundly capable of spawning religion but religion cannot legitimately deny evolution. Nature thus reigns supreme and we fantasize against it at our folly.

Resplendence: A Paradigm Shift from Religion to Reparadise the Earth
www.resplendence.org
Conclusion: The Tree of Life, the Selfish Gene, and Climax Genetic Diversity
The picture conveyed by the significance of endosymbiosis, genome fusion and horizontal transfer as key evolutionary processes complementing the vertical transmission of the tree of life, makes clear that evolution is not just a matter of competitive survival of the fittest gene, individual, or species, but of dynamic survival of genes in a surviving ecosystem. Although Dawkins' (1978) notion of the "selfish gene" was pivotal in drawing attention to the fact that it was the survival of genes and not organisms, or even species, that was the key evolutionary process, attributing the human sentiment of selfishness to a gene is somewhat of a self-serving advertising distraction on the part of the author, which diminishes the subtlety and complexity of the sometimes apparently paradoxical ways genes actually interact to bring about beneficial outcomes in the evolutionary dynamics of the ecosystem.
Although the idea of selection of genes has been pivotal in defining the need to consider evolutionarily stable strategies under genetic variation in ways which have been subsequently confirmed time and time again in situations such as the sexual genetics of social insects such as bees and ants, social selection is by no means ineffectual, or much of sociobiology, including the biological basis of morality as an extension of reciprocal altruism, would cease to exist.
Moreover, from what we have seen, particularly about horizontal gene transfer, and the capacity of mobile elements to induce modulated changes in nuclear genomes, it is not the 'selfishness' of a genetic element alone that results in survival of both a gene and its hosts, but dynamic feedbacks and relationships which ultimately contribute to a massive sharing of information in the manner of parallel genetic algorithms fundamental to the replicative genetic process, which enable global forms of genetic and genome optimization central to the overall viability of life as complex systems.

Fig 39: The Mandala of Evolution (Dion Wright) Click to see full image.
A predator, such as a lion survives, not because it is a selfish beast thinking only of eating the next gazelle, but because the predator, although it is surviving by killing individual antelopes, is maintaining a degree of stability in population dynamics, without which, the herbivores might multiply causing a massive famine, leading to cycles of boom and bust and the potential extinction or attrition of antelopes, lions and the grasslands.
Likewise, although we may think of individual genes, transposable elements, or viruses as 'selfish' for reproducing sufficiently to ensure their own survival, and sometimes behaving as noxious parasites, the overall effects of this process, in evolution can be to enrich the genetic potential of many unrelated organisms along the way, changing forever the face of the ecosystems in which they exist, enabling organisms of far greater complexity to evolve and to survive in the closing circle of the biosphere.
What humanity needs to learn to come to terms with is that our own survival is inextricably entwined with the survival of the immortal evolutionary tree tree of the diversity of life and it is this tree and the biosphere that contains it that we need to give our unswerving devotion to or we will fail the acid test of being a species co-dependent in a perennial ecosystem. By corrupting the tree of life through human impacts of climate change and habitat destruction we are invoking a mass extinction of life and may all too easily become one of the many casualties unless we care for the Tree as our primary goal in life.
References:








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
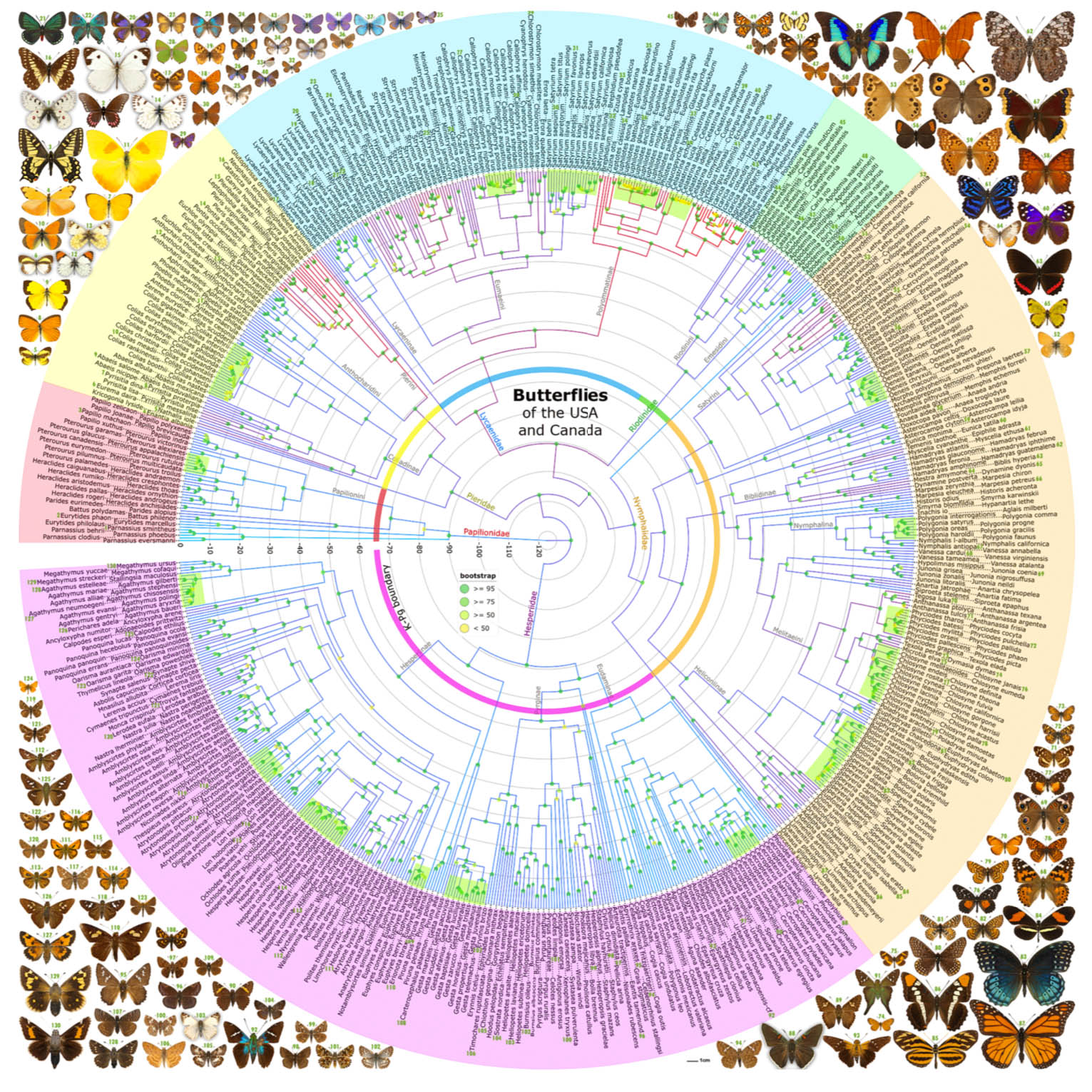{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}